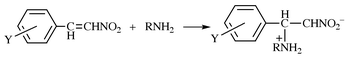
Kinetics and mechanism of the addition of benzylamines to β-nitrostyrenes in acetonitrile
Received (in Cambridge, UK) 16th August 1999, Accepted 14th October 1999
First published on UnassignedUnassigned23rd December 1999
Abstract
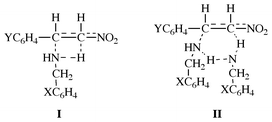
The kinetics and mechanism of the addition of benzylamines to β-nitrostyrenes in acetonitrile at 25.0 °C have been investigated. The addition reaction proceeds by two pathways, uncatalyzed (k2) and catalyzed (k3) paths. The kinetic isotope effects (kH/kD) involving deuterated benzylamine nucleophiles support the proposal that proton transfer from the amine to the β-carbon occurs concurrently with addition of the amine to the α-carbon. The transition state is predicted to have four- (I) and six-membered (II) cyclic structures for the k2 and k3 paths, respectively, with a tighter and more rigid structure for the uncatalyzed process. The cross-interaction constants, ρXY, are negative (−0.90 and −0.54 when the fall-off factor is taken into account) and the magnitude is larger for the uncatalyzed path than for the normal backside attack SN2 reaction of benzyl derivatives with anilines (−0.6 to −0.8).
Introduction
In our endeavors to establish the cross-interaction constants, ρij and βij in eqns. (1) and (2) (where i and j can be any two fragments X, Y or Z, involved in a transition state (TS), e.g. Scheme 1), as a mechanistic tool for organic reactions,1 we have shown that the sign of ρXY and βXY (Scheme 1) for the bond-making step is negative while that of ρYZ and βYZ for the bond-breaking step is positive in normal backside attack SN2 reactions.1,2 In general, the magnitude of ρij (and βij) represents a change in the strength of interaction between the two fragments, i and j, on going from the reactants to the TS.3 Thus the magnitude of ρXY is greater while that of ρYZ is smaller, when the TS is tighter with a larger degree of bond making (shorter rXY) and a smaller degree of bond cleavage (shorter rYZ).3| | | log (kij/kHH) = ρiσi + ρjσj + ρijσiσj |
(1)
|
| | | log (kij/kHH) = βipKi + βjpKj + βijpKipKj |
(2)
|
 |
| | Scheme 1 Typical SN2 transition state. R and σ represent reaction center and substituent on each fragment, r is the distance between fragments.
| |
In the limiting case in which only bond cleavage occurs in the TS, i.e., in the SN1 TS, ρYZ was indeed positive and large.4 In this work, we have studied the other limiting case, in which only bond formation takes place in the TS, using the nucleophilic addition reactions of benzylamines to (E![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) )-β-nitrostyrenes.
)-β-nitrostyrenes.
Although mechanistic studies of nucleophilic additions to an activated olefins including β-nitrostyrene have been reported,5 we have found no examples of reactions in which the structures (or substituents) of both the nucleophile (X) and the olefin (e.g. substituent Y in the β-nitrostyrene) are varied simultaneously.
We have carried out kinetic studies of addition reactions of benzylamines (BA) to (E)-β-nitrostyrenes (NS) in acetonitrile at 25.0 °C, eqn. (3). The main aim of this paper is to examine the substituent effects (both X and Y) on this addition reaction, and to determine the cross-interaction constant, ρXY, for the exclusive addition step in the TS. The sign and magnitude of ρXY are expected to provide a useful guide for predicting the TS structure.
| | 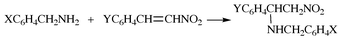 |
(3)
|
Results and discussion
The pseudo-first order rate constants (kobs) for all reactions studied in this work obeyed eqn. (4), indicating that the addition of BA to NS is catalyzed by a second BA molecule. The k2 (uncatalyzed) and k3 (catalyzed) values were determined as the intercept and slope, respectively, of a linear plot of kobs/[BA] vs. [BA]. The details of the experimental conditions, i.e., [BA], [NS], ranges of kobs and extinction coefficient changes at λmax, are given in the Experimental section (vide infra). The k2 and k3 values are summarized in Tables 1 and 2, where the Hammett (ρX and ρY) and Brønsted (βX) coefficients are also shown. Jalani et al. reported5c the k2 and k3 values with 28 X substituents but with Y = H only in eqn. (3). Comparison of their values at 25.0 °C with our k2 and k3 values in Tables 1 and 2 shows that our values are consistently greater, by a factor of 1.7. In their experimental section, they report that the kobs values were evaluated from the linear plot of log[NS] vs. time. The slope of this plot is actually smaller by a factor of 2.303 than the true kobs value, since kobs is the slope of a linear plot of ln[NS] (=2.303 log [NS]) vs. time.6 If we multiply their kobs value (and hence k2 and k3 values) by 2.303, our k2 and k3 values are consistently smaller by a factor of 1.2–1.3, which, we consider, is more appropriate since the difference in the temperature control may allow such small, but consistent, differences.| | | kobs = k2[BA] + k3[BA]2 |
(4)
|
Table 1 Second order rate constants (k2/10−2 dm3 mol−1 s−1) for the addition reactions of β-nitrostyrenes with X-benzylamines in acetonitrile at 25.0 °C. [NS] = 8.0 × 10−5 M and [BA] = 1.5–25 mM. ρX and ρY are the Hammett coefficients for X and Y. βX is the Brønsted coefficient for X
| | Y |
|---|
| X | p-Me | H | p-Cl | p-NO2 | ρYa |
|---|
| The σ values were taken from J. A. Dean, Handbook of Organic Chemistry, McGraw-Hill, New York, 1987, Table 7-1. Correlation coefficients were better than 0.998 in all cases. At 15 °C. At 5 °C. The σ values were taken from D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420. Correlation coefficients were better than 0.985 in all cases. Correlation coefficient was 0.997. The pKa values were taken from A. Fischer, W. J. Galloway and J. Vaughan, J. Chem. Soc., 1964, 3588. Correlation coefficients were better than 0.991 in all cases. X = p-CH3O were excluded from the Brønsted plot for βX (benzylamine) due to unreliable pKa value listed. |
|---|
| p-OMe | 2.55 | 4.46 | 15.2 | 128 | 1.82 ± 0.09 |
| | 2.40b | | | 118b | |
| | 2.24c | | | 108c | |
| | | | | | |
| p-Me | 1.92 | 3.86 | 11.9 | 86.2 | 1.74 ± 0.08 |
| | | | | | |
| H | 1.42 | 2.63 | 7.78 | 61.4 | 1.73 ± 0.06 |
| | | | | | |
| p-Cl | 0.649 | 1.12 | 3.02 | 20.6 | 1.60 ± 0.05 |
| | 0.605b | | | 18.9b | |
| | 0.562c | | | 17.1c | |
| | | | | | |
| ρXd | −1.17 ± 0.11 | −1.22 ± 0.15 | −1.41 ± 0.14 | −1.55 ± 0.19 | ρXYe = −0.41 |
| βXf | 1.20 ± 0.16 | 1.36 ± 0.14 | 1.51 ± 0.15 | 1.59 ± 0.29 | |
Table 2 Third order rate constants (k3/dm6 mol−2 s−1) for the addition reactions of β-nitrostyrenes with X-benzylamines in acetonitrile at 25.0 °C. [NS] = 8.0 × 10−5 M and [BA] = 1.5–25 mM. ρX and ρY are the Hammett coefficients for X and Y. βX is the Brønsted coefficient for X
| | Y |
|---|
| X | p-Me | H | p-Cl | p-NO2 | ρYa |
|---|
Same as the footnotes for Table 1, except correlation coefficients, r: r ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 0.994. Same as the footnotes for Table 1. Same as the footnotes for Table 1. Same as the footnotes for Table 1, except correlation coefficients, r: r 0.995. Same as the footnotes for Table 1, except correlation coefficients, r: r 0.996. Same as the footnotes for Table 1, except correlation coefficients, r: r 0.995. 0.994. Same as the footnotes for Table 1. Same as the footnotes for Table 1. Same as the footnotes for Table 1, except correlation coefficients, r: r 0.995. Same as the footnotes for Table 1, except correlation coefficients, r: r 0.996. Same as the footnotes for Table 1, except correlation coefficients, r: r 0.995. |
|---|
| p-OMe | 1.51 | 4.46 | 11.0 | 88.3 | 1.81 ± 0.13 |
| | 1.15b | | | 67.0b | |
| | 0.867c | | | 49.9c | |
| | | | | | |
| p-Me | 1.33 | 3.21 | 8.30 | 64.1 | 1.75 ± 0.08 |
| | | | | | |
| H | 0.833 | 2.33 | 5.84 | 40.3 | 1.72 ± 0.14 |
| | | | | | |
| p-Cl | 0.481 | 1.15 | 2.76 | 20.0 | 1.67 ± 0.08 |
| | 0.351b | | | 15.7b | |
| | 0.254c | | | 12.1c | |
| | | | | | |
| ρXd | −1.04 ± 0.06 | −1.15 ± 0.08 | −1.19 ± 0.08 | −1.29 ± 0.02 | ρXYe = −0.24 |
| βXf | 1.10 ± 0.08 | 1.13 ± 0.11 | 1.21 ± 0.11 | 1.26 ± 0.01 | |
The magnitudes of ρX for k2 (−1.2 to −1.6, which corresponds to −2.7 to −3.4 if the fall-off factor of 2.191a,7 is taken into account for the CH2 group between substituent X and the functional center N) and βX (1.2–1.6) are relatively large, compared to the corresponding values of ρX (−1.89 ± 0.13) and βX (0.68)8 for the addition of anilines.5b This is an indication of a greater degree of bond formation in the TS for the BA relative to aniline addition. The magnitude of ρY is also large (ρY = 1.6–1.8) suggesting that negative charge development at the Cα![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) Cβ moiety should be large, which is consistent with the tight bond formation predicted by the large magnitude of ρX and βX. In agreement with these trends, the cross-interaction constant, ρXY, is also large and negative (ρXY = −0.41, which corresponds to −0.90 when the fall-off factor is taken into account). The corresponding values for the normal backside attack SN2 reaction of benzyl derivatives with anilines are ca. −0.6 to −0.8.1,2
Cβ moiety should be large, which is consistent with the tight bond formation predicted by the large magnitude of ρX and βX. In agreement with these trends, the cross-interaction constant, ρXY, is also large and negative (ρXY = −0.41, which corresponds to −0.90 when the fall-off factor is taken into account). The corresponding values for the normal backside attack SN2 reaction of benzyl derivatives with anilines are ca. −0.6 to −0.8.1,2
Surprisingly, our k3 values give very similar, only slightly smaller, magnitudes of ρX, βX, ρY and ρXY (−0.24, corresponding to −0.54 taking into account fall-off) to those for aniline addition. This may be taken as evidence for a slightly looser bond-making structure in the catalyzed TS. This is supported by the smaller kH/kD (>1.0) values for the catalyzed (k3) process in Tables 3 and 4. The normal kinetic isotope effects (kH/kD > 1.0) involving deuterated benzylamines (XC6H4CH2ND2) provide evidence for partial N–H(D) bond cleavage in the TS.1b We presume that proton transfer to Cβ occurs concurrently with the Cα–N bond formation in the TS, I and II. In the k2 path, relatively greater stretching of the N–H bond is required than in the k3 path, so the kH/kD values are greater in general. This is consistent with a lesser degree of bond formation in the catalyzed path, as indicated by the smaller magnitude of the selectivity parameters, ρX, βX and ρXY. For both paths, the largest kH/kD value is obtained for the X = p-OMe and Y =
p-NO2 set. This is quite reasonable, since for this set of substituents the extent of bond formation in the TS is the greatest with the largest magnitude of ρY and ρX (βX) values (Tables 1 and 2).
Table 3 Kinetic isotope effects on the second-order rate constants for the reactions of β-nitrostyrenes with deuterated X-benzylamines in acetonitrile at 25.0 °Ca
| X | Y | kH/10−2 M−1 s−1 | kD/10−2 M−1 s−1 | kH/kD |
|---|
| Concentrations of NS and BA are the same as in Tables 1 and 2. Standard deviations. |
|---|
| p-OMe | p-Me | 2.55 (±0.03) | 0.992 (±0.002) | 2.57 ± 0.06b |
| p-OMe | H | 4.46 (±0.06) | 1.56 (±0.03) | 2.86 ± 0.06 |
| p-OMe | p-Cl | 15.2 (±0.2) | 5.18 (±0.06) | 2.93 ± 0.05 |
| p-OMe | p-NO2 | 128 (±3) | 41.5 (±0.1) | 3.08 ± 0.06 |
| | | | | |
| p-Cl | p-Me | 0.649 (±0.005) | 0.282 (±0.002) | 2.30 ± 0.02 |
| p-Cl | H | 1.12 (±0.02) | 0.420 (±0.006) | 2.67 ± 0.04 |
| p-Cl | p-Cl | 3.02 (±0.04) | 1.10 (±0.02) | 2.74 ± 0.06 |
| p-Cl | p-NO2 | 20.6 (±0.4) | 7.43 (±0.07) | 2.77 ± 0.06 |
Table 4 Kinetic isotope effects on the third-order rate constants for the reactions of β-nitrostyrenes with deuterated X-benzylamines in acetonitrile at 25.0 °Ca
| X | Y | kH/10−2 M−2 s−1 | kD/10−2 M−2 s−1 | kH/kD |
|---|
| Concentrations of NS and BA are the same as in Tables 1 and 2. Standard deviations. |
|---|
| p-OMe | p-Me | 1.51 (±0.04) | 1.17 (±0.02) | 1.29 ± 0.04a |
| p-OMe | H | 4.46 (±0.06) | 3.41 (± 0.05) | 1.31 ± 0.03 |
| p-OMe | p-Cl | 11.0 (±0.2) | 7.96 (±0.07) | 1.38 ± 0.03 |
| p-OMe | p-NO2 | 88.3 (±0.9) | 46.5 (±0.6) | 1.90 ± 0.03 |
| | | | | |
| p-Cl | p-Me | 0.481 (±0.004) | 0.333 (±0.003) | 1.44 ± 0.02 |
| p-Cl | H | 1.15 (±0.02) | 0.650 (±0.001) | 1.77 ± 0.04 |
| p-Cl | p-Cl | 2.76 (±0.05) | 1.50 (±0.02) | 1.84 ± 0.04 |
| p-Cl | p-NO2 | 20.0 (±0.3) | 10.6 (±0.2) | 1.87 ± 0.04 |
Finally, the activation parameters in Table 5 support the proposed mechanism. We note that in general the ΔH‡ values are higher but the negative ΔS‡ values are, in general, large. This is in accord with the greater degree of bond formation (exoergic) leading to less energy being required to break the π bond (endoergic) on CC but more rigid four-membered TS, I, for the k2 path. For the k3 path, the six-membered TS, II, has a looser (than I) structure with smaller negative ΔS‡.
Table 5 Activation parametersa for the reactions of β-nitrostyrenes with X-benzylamines in acetonitrile at 25.0 °Cb
| X | Y | Reactionpath | ΔH‡/kcalmol−1 | −ΔS‡/ calmol−1 K−1 | ΔG‡/kcalmol−1 |
|---|
| Calculated by the Eyring equation. The maximum errors calculated (by the method of K. B. Wiberg, Physical Organic Chemistry, Wiley, New York, 1964, p. 378) are ±0.5 kcal mol−1 and ±2 eu for ΔH‡ and ΔS‡, respectively. Concentrations of NS and BA are the same as in Tables 1 and 2. |
|---|
| p-OMe | p-Me | k2 | 0.5 | 64 | 19.6 |
| p-OMe | p-NO2 | k2 | 0.9 | 55 | 17.3 |
| p-Cl | p-Me | k2 | 0.5 | 61 | 18.7 |
| p-Cl | p-NO2 | k2 | 1.0 | 58 | 18.3 |
| | | | | | |
| p-OMe | p-Me | k3 | 4.0 | 44 | 17.1 |
| p-OMe | p-NO2 | k3 | 4.6 | 32 | 14.1 |
| p-Cl | p-Me | k3 | 4.6 | 44 | 17.7 |
| p-Cl | p-NO2 | k3 | 3.8 | 41 | 16.0 |
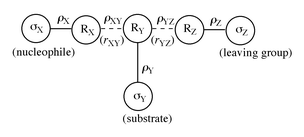
It is well known that there is a charge imbalance in the TS for the addition reactions of amines to activated olefins such as β-nitrostyrenes,5a,10eqn. (5). The TS “imbalance” was attributed to a lag in the charge delocalization into the NO2 moiety behind C–N bond formation in the TS.5b,10 This sort of imbalance leads to an extreme structure in which negative charge builds up on the Cβ carbon, as in III, instead of delocalizing onto the NO2 group, as in IV,10 in the TS. Since the negative charge transferred from the amine to the substrate is practically localized on Cβ in the TS, the proton transfer to Cβ from the amine should become viable, and our proposal of the proton transferred TS structures, I and II, is supported. The low cost of energy (ΔH‡) required for the π-bond cleavage in the TS may also be partly attributable to the concurrent proton transfer. In view of the results of Bernasconi et al.,5a that there is considerable positive charge development on the amine nitrogen in the TS, the positive charge carried away by the transferring proton should be small, i.e., the N⋯H bond stretching should be in its early stage in I and II.
| |  |
(5)
|
In summary, the addition of benzylamines to (E)-β-nitrostyrenes in acetonitrile proceeds by two pathways, the uncatalyzed (k2) and catalyzed (k3) paths. Furthermore, the reaction is a one-step process, in which the proton transfer from benzylamine to the β-carbon occurs concurrently with the addition of benzylamine to the α-carbon. This assertion is supported by the observation of relatively large primary kinetic isotope effects, kH/kD > 1.0, for deuterated nucleophiles, XC6H4CH2ND2. The charge imbalance leading to a practically localized negative charge on Cβ in the TS, III, seems to provide support for the proposed concurrent proton transfer. Bond formation in the TS is more advanced in the k2 (larger negative ρXY) than k3 path, so that the k2 path has a more rigid (more negative ΔS‡) four-membered structure with a greater degree of proton transfer.
Experimental
Materials
Solvent, acetonitrile (Merck GR) was used after three distillations. Benzylamines (Aldrich GR) were used after recrystallization. The β-nitrostyrenes were prepared by the literature method of Worrall.11 The analytical data are as follows (IR: NICOLET 5BX FTIR; NMR: JEOL 400 MHz).(E![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )-p-Methyl-β-nitrostyrene.. Mp 97–99 °C; λmax 324 nm; IR(KBr) νmax/cm−1 3100 (C–H, alkene), 1636 (CC, alkene), 1595 (CC, aromatic), 1431 (–CH2 bend); 1H NMR (400 MHz, CDCl3) 2.41 (3H, s, –CH3), 7.26 (2H, d, m-H, J 7.81 Hz), 7.44 (2H, d, o-H, J 8.31 Hz), 7.57 (1H, d, CH, J 13.7 Hz), 7.99 (1H, d, CH, J 13.7 Hz); 13C NMR (100.4 MHz, CDCl3) 143.1, 139.2, 136.3, 130.2, 129.2, 127.3, 100.6, 21.7.
)-p-Methyl-β-nitrostyrene.. Mp 97–99 °C; λmax 324 nm; IR(KBr) νmax/cm−1 3100 (C–H, alkene), 1636 (CC, alkene), 1595 (CC, aromatic), 1431 (–CH2 bend); 1H NMR (400 MHz, CDCl3) 2.41 (3H, s, –CH3), 7.26 (2H, d, m-H, J 7.81 Hz), 7.44 (2H, d, o-H, J 8.31 Hz), 7.57 (1H, d, CH, J 13.7 Hz), 7.99 (1H, d, CH, J 13.7 Hz); 13C NMR (100.4 MHz, CDCl3) 143.1, 139.2, 136.3, 130.2, 129.2, 127.3, 100.6, 21.7. (E)-β-Nitrostyrene.. Mp 58–60 °C; λmax 311 nm; IR(KBr) νmax/cm−1 3113 (C–H, alkene), 1629 (CC, alkene) 1582, 1475 (CC, aromatic), 1448 (–CH2 bend); 1H NMR (400 MHz, CDCl3) 7.45 (2H, d, m-H, J 6.84 Hz), 7.55 (2H, d, o-H) 7.59 (1H, d, CH, J 13.7 Hz), 8.01 (1H, d, CH, J 13.7 Hz); 13C NMR (100.4 MHz, CDCl3) 139.1, 137.1, 132.2, 130.1, 129.4, 129.2. (E)-p-Chloro-β-nitrostyrene.. Mp 102–104 °C; λmax 314 nm; IR(KBr) νmax/cm−1 3107 (C–H, alkene), 1636 (CC, alkene), 1589 (CC, aromatic), 1441 (–CH2 bend); 1H NMR (400 MHz, CDCl3), 7.43 (2H, d, m-H, J 8.79 Hz), 7.49 (2H, d, o-H, J 8.79 Hz), 7.57 (1H, d, CH, J 13.7 Hz), 7.97 (1H, d, CH, J 13.7 Hz); 13C NMR (100.4 MHz, CDCl3) 137.7, 137.4, 130.0, 129.8, 129.3, 128.5, 127.3. (E)-p-Nitro-β-nitrostyrene.. Mp 194–196 °C; λmax 307 nm; IR(KBr) νmax/cm−1 3101 (C–H, alkene), 1635 (CC, alkene), 1594 (CC, aromatic), 1431 (–CH2 bend); 1H NMR (400 MHz, CDCl3) 7.64 (1H, d, CH, J 13.7 Hz), 7.73 (2H, d, m-H, J 8.79 Hz), 8.04 (1H, d, CH, J 13.7 Hz), 8.32 (2H, d, o-H, J 8.79 Hz); 13C NMR (100.4 MHz, CDCl3) 136.7, 135.0, 129.7, 128.9, 128.3. Kinetic measurements
The reaction was followed spectrophotometrically by monitoring the decrease in the concentration of β-nitrostyrene, [NS], at λmax of the substrate to over 80% completion. The reaction was studied under pseudo-first-order conditions, [NS] = 8.0 × 10−5 M−1 and [BA] = (1.5–25) × 10−3 M−1 at 25.0 ± 0.1 °C. The pseudo-first-order rate constant, kobs, was determined from the slope of the plot (r > 0.993) ln[NS] (2.303 log [NS]) vs. time. The uncatalyzed (k2) and catalyzed (k3) rate constants were determined by fitting the kobs data to a parabolic curve, Fig. 1, of the kobsvs. [BA] plot, eqn. (4), which corresponded to a linear plot of kobs/[BA] vs. [BA] with an intercept of k2 and slope of k3. The two methods gave the same results within an experimental error of ±3%. The ranges of [BA] (1.5–25 mM] and the concentration of [NB] (8.0 × 10−5 M) were fixed in all cases. The ranges of kobs and extinction coefficients changes at λmax are summarized in Table 6. Representative data are shown in Table 7.
Table 6 The λmax (nm) ranges of extinction coefficient (ε/dm3 mol−1 cm−1) and (kobs/10−4 s−1) and percentage completion of reactions. [NS] = 8.0 × 10−5 M and [BA] = 1.5–25 mM
| X | Y | λmax/nm | ε/dm3 mol−1cm−1 | kobs/10−4s−1 | Completion (%) |
|---|
| p-OMe | p-Me | 324 | 1.60–0.20 | 0.406–15.5 | 87.5 |
| | H | 311 | 1.50–0.18 | 0.751–37.1 | 88.0 |
| | p-Cl | 314 | 1.70–0.21 | 2.49–108 | 87.6 |
| | p-NO2 | 307 | 1.90–0.23 | 15.5–878 | 88.4 |
| | | | | | |
| p-Me | p-Me | 324 | 1.80–0.21 | 0.364–13.5 | 88.3 |
| | H | 311 | 1.55–0.19 | 0.579–29.4 | 87.7 |
| | p-Cl | 314 | 1.75–0.22 | 1.88–82.0 | 87.4 |
| | p-NO2 | 307 | 1.90–0.25 | 14.9–637 | 86.8 |
| | | | | | |
| H | p-Me | 324 | 1.42–0.17 | 0.224–8.93 | 88.0 |
| | H | 311 | 1.50–0.15 | 0.344–20.1 | 90.0 |
| | p-Cl | 314 | 1.20–0.15 | 1.30–56.1 | 87.5 |
| | p-NO2 | 307 | 1.75–0.20 | 9.47–385 | 88.6 |
| | | | | | |
| p-Cl | p-Me | 324 | 1.50–0.18 | 0.108–4.83 | 88.0 |
| | H | 311 | 1.75–0.22 | 0.180–9.78 | 87.4 |
| | p-Cl | 314 | 1.25–0.15 | 0.283–22.8 | 88.0 |
| | p-NO2 | 307 | 1.65–0.20 | 3.02–195 | 87.8 |
Table 7 The kobsversus [BA] (benzylamine concentration/M) data in acetonitrile at 25.0 °C
| X | Y | [BA] M | kobs/10−4 s−1 | k2/10−2 M−1 s−1 | k3/M−2 s−1 | R corr. coeff. |
|---|
| p-OMe | p-Me | 0.0015 | 0.406 | 2.55 ± 0.04 | 1.51 ± 0.03 | 0.999 |
| | | 0.0030 | 0.975 | | | |
| | | 0.0045 | 1.47 | | | |
| | | 0.0060 | 1.96 | | | |
| | | 0.0075 | 2.74 | | | |
| | | 0.0100 | 4.03 | | | |
| | | 0.0150 | 7.34 | | | |
| | | 0.0200 | 11.32 | | | |
| | | 0.0250 | 15.55 | | | |
| | | | | | | |
| p-OMe | p-Cl | 0.0015 | 2.49 | 15.2 ± 0.3 | 11.0 ± 0.2 | 0.999 |
| | | 0.0030 | 5.76 | | | |
| | | 0.0045 | 9.21 | | | |
| | | 0.0060 | 13.3 | | | |
| | | 0.0075 | 17.3 | | | |
| | | 0.0100 | 25.6 | | | |
| | | 0.0150 | 47.1 | | | |
| | | 0.0200 | 74.7 | | | |
| | | 0.0250 | 108 | | | |
| | | | | | | |
| p-Me | p-Cl | 0.0015 | 1.88 | 11.9 ± 0.4 | 8.30 ± 0.03 | 0.994 |
| | | 0.0030 | 4.18 | | | |
| | | 0.0045 | 7.17 | | | |
| | | 0.0060 | 10.2 | | | |
| | | 0.0075 | 14.7 | | | |
| | | 0.0100 | 20.6 | | | |
| | | 0.0150 | 34.8 | | | |
| | | 0.0200 | 57.0 | | | |
| | | 0.0250 | 82.0 | | | |
| | | | | | | |
| p-Me | p-NO2 | 0.0015 | 14.9 | 86.2 ± 0.3 | 64.1 ± 0.2 | 0.996 |
| | | 0.0030 | 32.7 | | | |
| | | 0.0045 | 52.2 | | | |
| | | 0.0060 | 74.4 | | | |
| | | 0.0075 | 98.8 | | | |
| | | 0.0100 | 147 | | | |
| | | 0.0150 | 264 | | | |
| | | 0.0200 | 420 | | | |
| | | 0.0250 | 637 | | | |
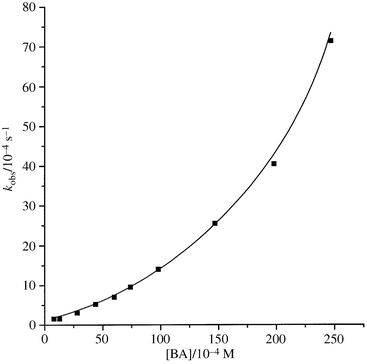 |
| | Fig. 1 The plot of kobsvs. concentration of benzylamine for the reaction of (E)-p-chloro-β-nitrostyrene in MeCN at 25.0 °C.
| |
Product analysis
β-Nitrostyrene (0.05 mol) and benzylamine (0.5 mol) were reacted in acetonitrile at 25.0 °C. After more than 15 half lives, solvent was removed under reduced pressure and product was separated by column chromatography (silica gel, 20% ethylacetate–n-hexane). Analytical data are as follows.C6H5CH(NHCH2C6H4OCH3)CH2NO2.. Liquid; IR(KBr) νmax/cm−1 2993 (C–H, CH2), 2958 (N–H, stretch), 2946 (C–H, CH3), 1460 (CC, aromatic), 1448 (C–H, CH2); 1H NMR (400 MHz, CDCl3) 3.82 (3H, s, OCH3), 4.30 (1H, t, CH, J 6.90 Hz), 4.76 (2H, d, benzylamine CH2, J 3.01 Hz), 4.78 (2H, d, benzyl CH2, J 6.10 Hz), 6.88–7.53 (9H, m, aromatic ring); 13C NMR (100.4 MHz, CDCl3) 134.0 130.7 129.4 128.9, 128.6, 128.5, 128.3, 127.2, 114.1, 113.8, 64.9, 55.2, 41.7. Acknowledgements
The authors wish to acknowledge the financial support of the Korea Research Foundation made in the program year of 1998.References
-
(a) I. Lee, Adv. Phys. Org. Chem., 1992, 27, 57 Search PubMed;
(b) I. Lee, Chem. Soc. Rev., 1995, 24, 223 RSC.
- I. Lee, C. S. Shim, S. Y. Chung, H. Y. Kim and H. W. Lee, J. Chem. Soc., Perkin Trans. 2, 1988, 1919 RSC.
- I. Lee, J. Phys. Org. Chem., 1992, 5, 736 CAS.
- I. Lee, M. S. Choi and H. W. Lee, J. Chem. Res. (S), 1994, 92 Search PubMed; I. Lee, M. S. Choi and H. W. Lee, J. Chem. Res. (M), 1994, 568 Search PubMed.
-
(a) C. F. Bernasconi, R. A. Renfrow and P. R. Tia, J. Am. Chem. Soc., 1986, 108, 4541 CrossRef CAS;
(b) B. Varghese, S. Kothari and K. K. Banerji, J. Chem. Res. (S), 1998, 422 RSCB. Varghese, S. Kothari and K. K. Banerji, J. Chem. Res. (M), 1998, 1853 Search PubMed;
(c) N. Jalani, S. Kothari and K. K. Baneryi, Can. J. Chem., 1996, 74, 625 CAS.
- F. Ruff
and I. G. Csizmadia
, Organic Reactions. Equilibria, Kinetics and Mechanism, Elsevier, Amsterdam, 1994, p. 62.
Search PubMed.
- C. Hansch, D. Hoekman and H. Gao, Chem. Rev., 1996, 96, 1045 CrossRef CAS.
- Estimated by ρX/ρe = βX,1a where ρe is the Hammett coefficient for dissociation of anilinium ions (pKa) vs.σ, which is ρe = 2.77.9.
- C. D. Johnson
, The Hammett Equation, Cambridge University Press, Cambridge, 1993, p. 30.
Search PubMed.
-
(a) C. F. Bernasconi, Acc. Chem. Res., 1987, 20, 301 CrossRef CAS;
(b) C. F. Bernasconi, Tetrahedron, 1989, 45, 4017 CrossRef CAS.
- D. E. Worrall
, in Organic Synthesis, H. Gilman and A. Blatt, eds., Wiley, New York, 1941, Coll. Vol. 1, p. 413.
Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.