Flash photolysis investigation of the reaction of phenylselanyl radicals with hexabutyldistannane
Irina P.
Beletskaya
*a,
Alexander S.
Sigeev
a,
Vladimir A.
Kuzmin
b,
Alexander S.
Tatikolov
b and
Laszlo
Hevesi
c
aDepartment of Chemistry, Moscow State University, GSP-3, Moscow, 119899, Russia. Fax: 7(095)938-1844; E-mail: beletska@org.chem.msu.su
bN. M. Emanuel Institute of Biochemical Physics, Russian Academy of Sciences, Kosygina str, 4, 117977, Moscow, Russia
cDepartment de Chemie, Facultes Universitaires Notre-Dame de la Paix, Rue de Bruxelles, 61, B-500, Belgium. Fax: +32 (81) 72-45-30; E-mail: laszlo.hevesi@fundp.ac.be
First published on 25th January 2000
Abstract
The reaction of the phenylselanyl radical with hexabutyldistannane was studied by flash photolysis and the rate constants and activation parameters were determined.
The generation and reactivity of selenium-centered organoselenium radicals attracts much attention because these radicals are usually formed upon photolysis of a variety of organoselenium compounds.1 In particular, the formation of phenylselanyl radical, and its reactions with tertiary phosphines,2 activated alkenes,3 and alkynes,4 have been well studied. The recombination kinetics of PhSe· has been investigated and the rate constants for its reactions with a number of vinyl monomers have been measured using flash photolysis techniques.3a
It has been shown earlier that irradiation of a mixture of the diselenides R2Se2 and R′2Se2 leads to an equilibrium mixture which contains the mixed diselenide RSeSeR′ formed upon recombination of the corresponding radicals.5 Similarly, tin selenides R3SnSePh are formed upon irradiation of a mixture of diphenyl diselenide and hexaalkyldistannanes.6 Unlike the exchange reaction between two diselenides, this process can serve as a convenient method for the synthesis of the exchange product.
In this work we have studied the reactions of the phenylselanyl radical obtained upon photolysis of Ph2Se2 with UV light in the 310–370 nm range. Flash photolysis of oxygen-free and air-saturated solutions of Ph2Se2 (10−3 M) in toluene, hexane, and CCl4 leads to formation of the phenylselanyl radical according to eqn. (1).3a
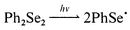
| (1) |
The absorption spectra of PhSe· recorded in this work and its rate of recombination, measured at 490 nm in hexane and CCl4, are the same or similar to those described earlier![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 3a (Fig. 1). The decay of the radicals obeys a second-order law according to the recombination reaction (2).
3a (Fig. 1). The decay of the radicals obeys a second-order law according to the recombination reaction (2).
![Spectrum of phenylselanyl radical in hexane. [PhSeSePh] = 2.8 × 10−4 M (•, 32; ■, 60; ▲, 100; ×, 260 ms after flash).](/image/article/2000/P2/a906072c/a906072c-f1.gif) | ||
| Fig. 1 Spectrum of phenylselanyl radical in hexane. [PhSeSePh] = 2.8 × 10−4 M (•, 32; ■, 60; ▲, 100; ×, 260 ms after flash). | ||
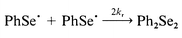
| (2) |
The decay kinetics of the radicals corresponds to the value of 2kr/ε (485 nm) = 8 × 106 cm s−1 in hexane. According to Ito,3a the diffusion controlled reaction with the rate constant 2kr = 7 × 109 M−1 s−1 corresponds to decay of the radicals. Under the experimental conditions, [PhSe·] ≈ 10−6 M; therefore, τ1/2 = 1/(2kr[PhSe·]) ≈ 200 μs. Oxygen has essentially no effect on the decay kinetics of phenylselanyl radicals.
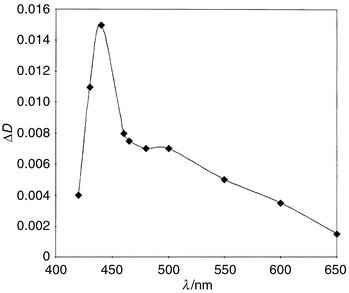
Flash irradiation of Bu6Sn2 through UFS-6 light filters (320–390 nm) both in the presence and in the absence of oxygen did not lead to formation of any intermediates. Only in the case of flash irradiation of oxygen-free Bu6Sn2 solutions with unfiltered light (UV light with wavelength down to 220 nm), the absorption spectrum of a short-lived transient with λmax = 440 nm can be observed (Fig. 2). This can apparently be assigned to the tributylstannyl radical, eqn. (3).
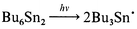
| (3) |
The decay kinetics of these radicals obeys a second-order law (with the value of 2k1/ε = 3 × 106 cm s−1, eqn. (4)).
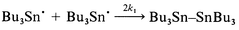
| (4) |
Tributylstannyl radicals efficiently react with oxygen in air-saturated solutions (k2 ≈ kdiff ≈ 109 M−1 s−1) according to eqn. (5).7
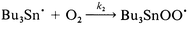
| (5) |
In this work the radical substitution reaction (6) with hexabutyldistannane was studied in toluene and acetone using flash photolysis techniques.

| (6) |
Since the tributylstannyl radical formed efficiently reacts with oxygen in air, other reactions of this radical (e.g., with Ph2Se2 present in the system) are excluded in air-saturated solutions. It is experimentally confirmed in this work that in air-saturated solutions and in the presence of sufficiently high Bu6Sn2 concentration (>10−4 M; [Bu6Sn2] ≫ [PhSe·]) the decay reaction of phenylselanyl radicals follows a pseudo-first-order equation d(PhSe·)/dt = keff[PhSe·], where keff = k3[Bu6Sn2]. Fig. 3 shows the plot used to obtain k3 = 1.5 × 106 M−1 s−1 in toluene. In acetone k3 = 3.0 × 106 M−1 s−1.
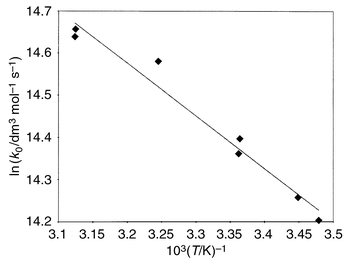 | ||
| Fig. 3 Calculation of the rate constant for the reaction of phenylselanyl radical with hexabutyldistannane in toluene. | ||
The temperature dependence of the reaction rate was measured in toluene in the temperature range of 13–50 °C (Fig. 4), and activation parameters were determined: Ea = 3.0 ± 0.5 kcal mol−1, A0 = 2.8 × 108 M−1 s−1. Such a low value of the activation energy and low pre-exponential factor might indicate a diffusion controlled reaction and strong steric effects, respectively. Note the important role of aerial oxygen in the interaction of phenylselanyl radical with hexabutyldistannane. A decrease in the lifetime of phenylselanyl radical in the presence of hexabutyldistannane according to the pseudo-first-order reaction was only observed in the presence of oxygen. In the absence of oxygen, the decay kinetics of the phenylselanyl radicals was essentially independent of the presence of hexabutyldistannane in solution up to [Bu6Sn2] = 10−2 M. Hence, in the absence of oxygen, the lifetime of tributylstannyl radical formed in reaction (6) is sufficiently long (tens of milliseconds), that its decay can occur through recombination (homo- and cross-recombination, eqns. (4) and (7), respectively) and through reaction with Ph2Se2, eqn. (8).
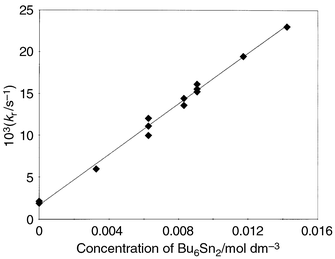 | ||
| Fig. 4 Calculation of the activation energy for the reaction of phenylselanyl radical with hexabutyldistannane in toluene. | ||
| Bu3Sn· + PhSe· → Bu3SnSePh | (7) |
| Bu3Sn· + Ph2Se2 → Bu3SnSePh + PhSe· | (8) |
Phenylselanyl radicals formed in reaction (8) can participate in the chain propagation reaction (6). Homo- and cross-recombination reactions (2), (4) and (7) are the chain termination steps.
We have also investigated the reactivity of phenylselanyl radicals towards phenylacetylene and diphenylacetylene, eqn. (9).
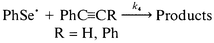
| (9) |
There is no change in the decay kinetics of the PhSe· radical in the presence of diphenylacetylene up to a concentration of 0.8 M which indicates the absence of reaction (9) for diphenylacetylene (in this case k4 ≪ 102 M−1 s−1). At the same time, the decay rate of PhSe· was accelerated in the presence of phenylacetylene (Fig. 5). The rate constant for the interaction of PhSe· with PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH was obtained from the dependence of the decay rate of PhSe· on phenylacetylene concentration (Fig. 5): k4 = 1.9 × 105 M−1 s−1. Hence, in agreement with ref. 4(b), PhSe· does not react with diphenylacetylene but reacts with phenylacetylene. The latter reaction leads to the formation of a photo-addition product mixture in which the E isomer predominates.4a
CH was obtained from the dependence of the decay rate of PhSe· on phenylacetylene concentration (Fig. 5): k4 = 1.9 × 105 M−1 s−1. Hence, in agreement with ref. 4(b), PhSe· does not react with diphenylacetylene but reacts with phenylacetylene. The latter reaction leads to the formation of a photo-addition product mixture in which the E isomer predominates.4a
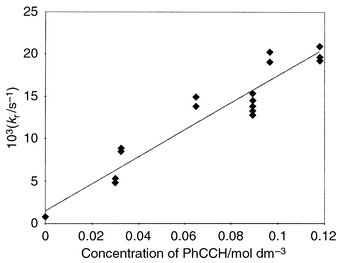 | ||
| Fig. 5 Calculation of the rate constant for the reaction of phenylselanyl radical with phenylacetylene. | ||
Conclusion
We have studied the photochemical generation of the phenylselenium radical in the presence of hexabutyldistannane and measured the rate of reaction leading to formation of Bu3SnSePh. Without O2 a chain process takes place. The stage of chain termination involves the homo- and hetero-recombination of PhSe· and Bu3Sn· radicals. In the presence of air O2 we did not observe a chain process due to the reaction of Bu3Sn· radical with O2.Experimental
Diphenyl diselenide was a gift from Ms S. V. Amosova and Mr V. A. Potapov and purified by recrystallization. Solvents were of spectrophotometric grade. Hexabutyldistannane and phenylacetylene were used after distillation at reduced pressure. Commercially available diphenylacetylene was purified by recrystallization.The flash photolysis experiments were carried out in a temperature-controlled quartz cell. The apparatus was of standard design.8
The flash energy of the xenon flash lamp was 50 J. The flash duration τ1/2 ≈ 7 μs. Light in the range of 310–370 nm was selected by the use of UFS-6 light filters. Kinetic observations were made with a continuous monitor light source and photomultiplier detection. The error in the determination of rate constants was ca. 15%. All solutions were degassed before photochemical measurements.
Acknowledgements
We are grateful to Ms S. V. Amosova and Mr V. A. Potapov for a gift of Ph2Se2. Support of this work by grant INTAS-RFBR No 95-0126 is gratefully acknowledged. We thank Professor A. Pelter for correction of the English.References
- E. N. Deryagina, M. G. Voronkov and N. A. Korchevin, Usp. Khim. (Russ. Chem. Rev.), 1993, 62, 1173 Search PubMed; L. Castle and M. J. Perkins , in The Chemistry of Organic Selenium and Tellurium Compounds, ed. S. Patai, Wiley Interscience, New York, 1987, vol. 2, p. 657; Search PubMed; U. Shmidt, A. Muller and K. Marakau, Chem. Ber., 1964, 97, 405 Search PubMed.
- D. H. Brown, R. J. Cross and D. Millington, J. Chem. Soc., Dalton Trans., 1977, 159 RSC.
- (a) O. Ito, J. Am. Chem. Soc., 1983, 105, 850 CrossRef CAS; (b) J. C. Scaiano and K. U. Ingold, J. Am. Chem. Soc., 1977, 99, 2079 CrossRef CAS; (c) G. A. Russell and J. Hershberg, J. Am. Chem. Soc., 1980, 102, 7603 CrossRef CAS.
- (a) T. G. Back and M. V. Krishna, J. Org. Chem., 1988, 53, 2533 CrossRef CAS; (b) A. Ogawa, H. Yokoyama, K. Yokoyama, T. Masawaki, N. Kambe and N. Sonoda, J. Org. Chem., 1991, 56, 5721 CrossRef CAS.
- V. A. Potapov, S. V. Amosova, P. A. Petrov, L. S. Romanenko and V. V. Keiko, Sulfur Lett., 1992, 15, 121 Search PubMed.
- A. S. Peregudov , A. S. Sigeev , B. A. Lukovskii , N. A. Donskaya and I. P. Beletskaya , Abstracts of the XIIth FECHEM Conference on Organometallic Chemistry, Prague, 1997, p. 175. Search PubMed.
- J. A. Howard, J. C. Tait and S. B. Tong, Can. J. Chem., 1979, 57, 2761 CAS.
- Yu. E. Borisevich, A. S. Tatikolov and V. A. Kuzmin, Khim. Vys. Energ. (High Energy Chem. (Russ.)), 1978, 5, 474 Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |