Chiral discrimination of 2-arylalkanoic acids by (1S,2R)-1-aminoindan-2-ol through the formation of a consistent columnar supramolecular hydrogen-bond network†
Kazushi Kinbara, Yuka Kobayashi and Kazuhiko Saigo*
Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
First published on UnassignedUnassigned23rd December 1999
Abstract
Enantiopure cis-1-aminoindan-2-ol was selected as a basic resolving agent for racemic 2-arylalkanoic acids on the basis that its rigid cis-conformation would favor the formation of a supramolecular hydrogen-bonded column, in which chiral discrimination of the racemic carboxylate would occur. It was found that this amino alcohol possesses high resolving efficiency for a variety of racemic acids; also, X-ray crystallographic analyses of the diastereomeric salts showed that a columnar hydrogen-bond network is formed in both the less- and more-soluble diastereomeric salts, as we had expected. A detailed study on the stabilising interactions suggested that there are two that play an important role: (i) hydrogen bonding between the ammonium and hydroxy groups and the acid carboxylate, which determines the formation of the columnar network and (ii) CH![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ⋯π, which influences the herringbone packing of the aromatic groups, implying that it also plays some role in chiral discrimination.
⋯π, which influences the herringbone packing of the aromatic groups, implying that it also plays some role in chiral discrimination.
Optical resolution of racemates via the formation of diastereomers with a resolving agent is a popular method for the preparation of enantiopure compounds.1 Unfortunately the variety of readily available resolving agents is limited, and resolution is sometimes not possible by using these resolving agents. The development of novel resolving agents requires us to predict a structure suitable for a given target racemate.2 The difficulty in this arises from our poor understanding of the relationship between the physical properties of pairs of diastereomeric salts and the characteristics of their molecular and/or crystal structure.
We have recently carried out the resolution of a series of racemic 2-arylalkanoic acids by enantiopure erythro-2-amino-1,2-diphenylethanol (1) (Fig. 1). We found that enantiopure 1 distinguished between the enantiomers of several kinds of 2-arylalkanoic acids.3 Where the resolution was efficient a supramolecular hydrogen-bonded column was formed. A network of strong hydrogen bonds between the carboxylate oxygens, the ammonium hydrogens of the protonated 1 cation (1·H+), the hydroxy group of 1·H+, and a water molecule, was commonly formed in the less-soluble diastereomeric salts (Fig. 2), while in the more-soluble diastereomeric salts another columnar network of hydrogen bonds between only the carboxylate oxygens and the ammonium hydrogens of 1·H+ was formed.
 | ||
| Fig. 1 Structures of resolving agents. | ||
 | ||
| Fig. 2 Schematic representation of the columnar hydrogen-bond networks formed in the less-soluble diastereomeric salts of enantiopure 1 with 2-arylalkanoic acids. The hydrogen atoms, and aryl and alkyl substituents are omitted for clarity. The dotted lines show the hydrogen bonds. | ||
No such stable structure was formed where resolution was poor. The different hydrogen-bond networks of the diastereomeric salts are thought to arise from the free rotation around the C1–C2 single bond and the steric repulsion between the two phenyl groups in 1, resulting in small differences in stability for less- and more-soluble diastereomeric salts in some cases.
On the basis of these results, we sought a resolving agent for racemic 2-arylalkanoic acids more suitable than 1; the fundamental principle for our selection is based on the hypothesis that the formation of a consistent columnar supramolecular hydrogen-bond network in the less-soluble diastereomeric salts is essential in order to distinguish the enantiomers of the racemic acids. First of all, in order to prevent the free rotation observed for 1·H+, we wanted a cyclic amino alcohol. Moreover, to mimic the gauche conformation of the ammonium and hydroxy groups of 1·H+ in crystals, which enables the formation of the hydrogen-bonded columns, a cyclic cis-amino alcohol was preferred. We finally chose enantiopure cis-1-aminoindan-2-ol (2) (Fig. 1) as it not only fulfilled the structural requirements, but is also readily available, and has been used in the resolution of some profen type drugs.4
In the present paper, we report the diastereomeric salt resolution of racemic 2-arylalkanoic acids by 2 and the chiral discrimination mechanism on the basis of the X-ray crystallographic analyses of the resultant diastereomeric salts.
Results and discussion
Resolution of 2-arylalkanoic acids with (1S,2R)-1-aminoindan-2-ol
Resolution of racemic 2-arylalkanoic acids with (1S,2R)-2 was performed under similar conditions to those reported in our previous paper.3 In order to make the crystallisation conditions as similar as possible, the resolutions were carried out upon single-crystallisation of an equimolar mixture of the corresponding racemic acid and (1S,2R)-2 from an alcohol–water mixture at 30 °C. Only when the salt did not crystallise from this solvent system was ethyl acetate used as a poor solvent. The amount of solvent was controlled so that the resulting diastereomeric salt was obtained as close as possible to the range of 50–90% yield (based on a half amount of the racemic acid to be resolved). The ratio of the solvents was controlled only to adjust the solubility of the salt, and had essentially little effect on the diastereomeric ratio of the precipitated salt; fine tuning of the resolution conditions was not carried out.5 The details of the conditions are described in the Experimental section.As can be seen from Table 1, (1S,2R)-2 showed high resolving efficiencies for a variety of racemic 2-arylalkanoic acids having a substituent on the aromatic ring and/or at the α-position. It is noteworthy that (1S,2R)-2 showed high resolving efficiency even for p-substituted 2-arylalkanoic acids, which could not be efficiently resolved by enantiopure 13 (Table 1, entries 4, 5, 8).
a
| ||||||
|---|---|---|---|---|---|---|
| Entry | Racemic acid | Solvent (ratiob) | Yield (%)c | Ee (%) | Resolving efficiencyg | Absolute configuration of major enantiomer |
| a The resolution was carried out on a 1–3 mmol scale. b The weight (g) of the solvent normalized to 1 mmol scale. c Yield of the crystallised diastereomeric salt based on a half amount of the racemic acid. d Enantiomeric excess (ee) of the liberated acid, as determined by HPLC analysis (Daicel Chiralcel OJ-R). e Ee of the liberated acid, as determined by HPLC analysis [Daicel Chiralcel OD (entries 3, 4, 6, 8, 10 and 12) and OB (entry 2)] after conversion into the methyl ester by diazomethane. f Ee of the liberated acid, as determined by a chiral GC (CHIRALDEX GT-A) after conversion into the methyl ester by diazomethane. g Defined as the product of the yield of the diastereomeric salt and the ee. | ||||||
| 1 | 3a | H2O–MeOH (0.2/0.7) | 65 | 95d | 0.62 | R |
| 2 | 3b | H2O–EtOH (0.5/0.1) | 85 | 24e | 0.21 | R |
| 3 | 3c | H2O–EtOH (0.2/0.8) | 59 | 91e | 0.54 | R |
| 4 | 3d | H2O–PriOH (0.2/2.3) | 32 | 61e | 0.20 | R |
| 5 | 3e | H2O–EtOH (0.2/0.4) | 79 | 88d | 0.70 | R |
| 6 | 3f | AcOEt (12.2) | 54 | 87e | 0.47 | R |
| 7 | 4a | H2O–EtOH (0.2/0.2) | 48 | 78d | 0.37 | R |
| 8 | 4b | H2O–EtOH (0.3/0.8) | 83 | 67e | 0.56 | R |
| 9 | 5 | H2O–EtOH (0.2/0.9) | 80 | 44d | 0.35 | S |
| 10 | 6 | H2O–EtOH (0.1/0.2) | 67 | 59e | 0.40 | S |
| 11 | 7 | AcOEt–EtOH (5.3/3.6) | 45 | 88f | 0.40 | S |

Here, it should be pointed out that the absolute configuration of the acid component in the less-soluble diastereomeric salts differed depending on the bulkiness of the alkyl group at the α-position of the 2-arylalkanoic acid; the (R)-form was predominantly obtained from 3a and 4a, the (S)-form from 5 and 6 (vide post).
Crystal structures of the less-soluble diastereomeric salts (α-alkyl group = Me, Et)
The lattice constants and other parameters obtained by the X-ray crystallographic analyses performed in the present study are summarised in Table 2.8| Compound | (1S,2R)-2·(R)-3a (less soluble) | (1S,2R)-2·(R)-3c (less soluble) | (1S,2R)-2·(R)-3e·H2O (less soluble) | (1S,2R)-2·(R)-4a (less soluble) | (1S,2R)-2·(S)-4a (more soluble) | (1S,2R)-2·(S)-4b (less soluble) | (1S,2R)-2·(S)-6 (less soluble) | (1S,2R)-2·(S)-7 (less soluble) |
|---|---|---|---|---|---|---|---|---|
| a |Fo| > 0.1σ|Fo|. | ||||||||
| Formula | C18H21NO3 | C19H23NO3 | C19H25NO5 | C19H23NO3 | C19H23NO3 | C20H25NO3 | C20H25NO3 | C19H20NO4F3 |
| Formula weight | 299.0 | 313.0 | 347.4 | 313.0 | 313.0 | 327.4 | 327.4 | 383.4 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21 | P21 | P21 | P21 | P21 | C2 | C2 | P2i |
| a/Å | 12.121(4) | 12.294(5) | 14.584(4) | 12.351(3) | 16.848(4) | 17.045(4) | 19.831(4) | 15.787(2) |
| b/Å | 6.319(2) | 6.388(4) | 6.175(3) | 6.170(2) | 5.947(2) | 6.148(2) | 5.786(1) | 6.3359(8) |
| c/Å | 10.882(4) | 11.134(5) | 10.852(3) | 11.431(2) | 8.580(2) | 17.931(2) | 16.453(2) | 9.353(1) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 102.69(3) | 103.71(3) | 107.80(2) | 99.10(2) | 101.75(2) | 101.22(2) | 110.57(1) | 99.428(8) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 813.1(5) | 849.5(7) | 930.6(6) | 860.2(8) | 841.7(4) | 1843.1(9) | 1767.7(5) | 922.9(2) |
| Z | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 2 |
| μ/mm−1 | 0.668 | 0.661 | 0.735 | 0.653 | 0.667 | 0.629 | 0.656 | 0.998 |
| Temperature/K | 293 | 293 | 293 | 293 | 293 | 293 | 293 | 293 |
| No. reflections measured | 1454 | 1288 | 1647 | 1474 | 1466 | 1648 | 1598 | 1613 |
| No. reflections observed (|Fo| > 3σ|Fo|) | 1403 | 1260a | 1563 | 1311 | 1387 | 1571 | 1540 | 1487 |
| Rint | 0.022 | 0.014 | 0.014 | 0.041 | 0.063 | 0.015 | 0.021 | 0.015 |
| R | 0.061 | 0.086 | 0.045 | 0.043 | 0.101 | 0.049 | 0.050 | 0.047 |
In all of the less- and more-soluble diastereomeric salts, a columnar supramolecular hydrogen-bond network is formed around the 21-axis, which is parallel to the b-axis. In addition, except for (1S,2R)-2·(R)-3e·H2O, no water molecules are incorporated in the crystals. These facts indicate that the cis-conformation of the ammonium and hydroxy groups of the protonated 2 cation (2·H+) causes the formation of a tight hydrogen-bonded column without hydrogen bonds with a water molecule, just as we had anticipated.
A common columnar hydrogen-bonded network is found in the less-soluble diastereomeric salts ((1S,2R)-2·(R)-3a, (1S,2R)-2·(R)-3c, (1S,2R)-2·(R)-3e·H2O, (1S,2R)-2·(R)-4a, and (1S,2R)-2·(R)-4b)) of which the acid part has methyl or ethyl group as the α-alkyl group. Their crystal structures and a schematic representation of the common hydrogen-bond network are shown in Figs. 3 and 4. It can be seen that the carboxylate oxygens, the ammonium hydrogens of 2·H+, and the hydroxy group of 2·H+ participate in the formation of the hydrogen-bond column, which is constructed around the 21-axis. The ammonium group of a crystallographically independent 2·H+ (Fig. 4, in the square) has three hydrogens, and two of them form hydrogen bonds with the oxygens of two different carboxylates (Fig. 4, A and B). These carboxylates are related to each other by translation along the b-axis. The other ammonium hydrogen of 2·H+ forms a hydrogen bond with the hydroxy oxygen of another 2·H+ (Fig. 4, C), which is related by the 21-axis (the columnar axis) to the original 2·H+ molecule. The hydroxy hydrogen of 2·H+ is used to form a hydrogen bond with the carboxylate oxygen (Fig. 4, D). This carboxylate is related by the 21-axis to the carboxylate hydrogen-bonded with the ammonium hydrogen. This pattern of hydrogen bonds is different from that usually found in the salts of carboxylic acids with primary amines.9 Here, the hydroxy group acts both as hydrogen donor and acceptor, and deeply penetrates into the columnar structure so as to form a novel hydrogen-bonding pattern.
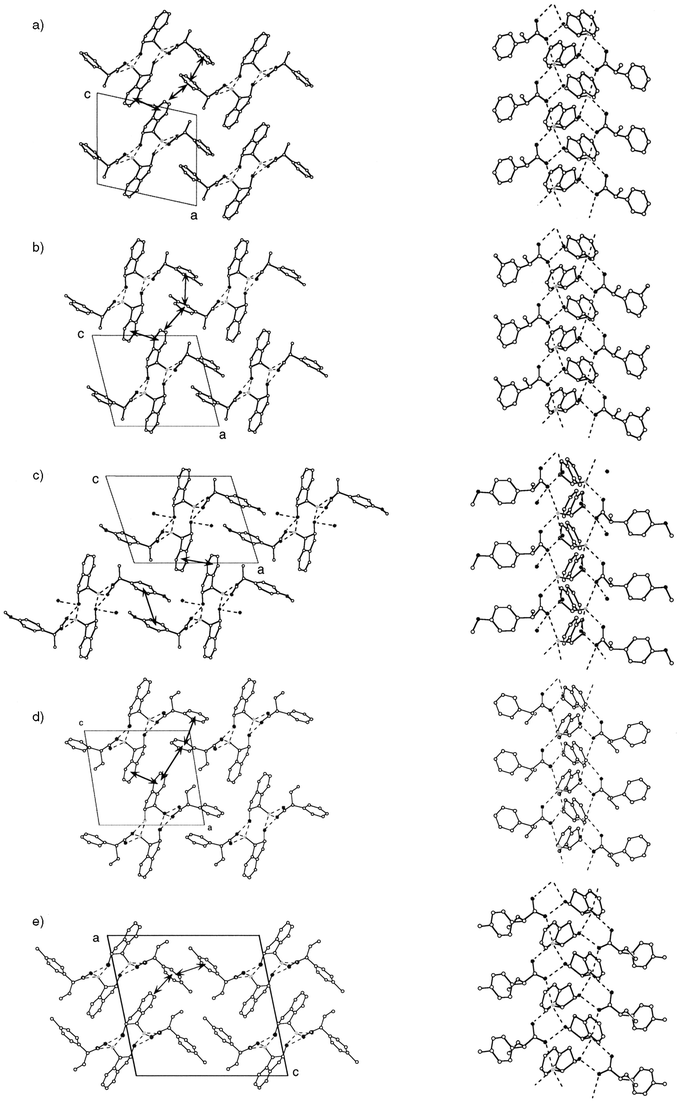 | ||
| Fig. 3 Crystal structures of the less-soluble diastereomeric salts viewed down the b-axis (left) and columnar hydrogen-bond network formed around the 21-axis (right). The hydrogen atoms are omitted for clarity. The dotted lines and arrows show the hydrogen bonds and herringbone packings, respectively. The solid square represents the unit cell. a) (1S,2R)-2·(R)-3a. b) (1S,2R)-2·(R)-3c. c) (1S,2R)-2·(R)-3e·H2O. d) (1S,2R)-2·(R)-4a. e) (1S,2R)-2·(R)-4b. | ||
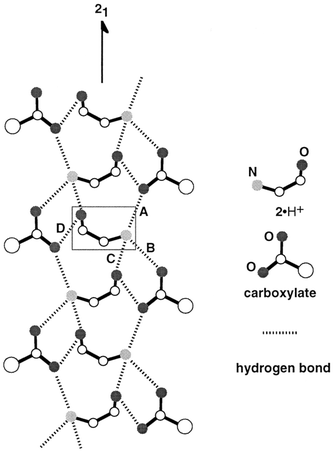 | ||
| Fig. 4 Schematic representation of the columnar hydrogen-bond network formed in the less-soluble diastereomeric salts. Hydrogen atoms as well as aryl and alkyl groups are omitted for clarity. The dotted lines show the hydrogen bonds. | ||
The intermolecular distances between N⋯O and O⋯O are 2.6–2.9 Å in most cases;10 these values indicate quite strong hydrogen bonding. In addition, the hydrogen bonds have two characteristics: 1) The ammonium and hydroxy groups are located on almost the same plane as the carboxylate groups, which is perpendicular to the ac-plane. 2) The hydrogens are directed towards the lone pairs of the carboxylate oxygens; this location of the hydrogens favors formation of strong hydrogen bonds.11
In the hydrogen-bond column, the smallest α-substituent of the carboxylate, the hydrogen, is commonly oriented parallel to the columnar axis, while the other two substituents, the aryl and alkyl groups, are positioned perpendicular to it. Moreover, the largest aryl group is located away from the aromatic group of 2·H+, while the medium alkyl group is close to 2·H+.
The aryl groups (Ar(2)) in two translational 2·H+ molecules in a single hydrogen-bond column, as well as those (Ar(acid)) in two translational carboxylate molecules, stack to each other along the columnar axis, i.e. the b-axis. The mode of stacking is close to that reported for stacked aromatic molecules in a crystal.12,13
The stacked Ar(2) and Ar(acid) form herringbone packings; two or three kinds of herringbone packings are found in the less-soluble salts (indicated by arrows in Fig. 3). Hence, it is suggested that CH⋯π interactions between these aromatic groups also play an important role in the stabilisation of these crystals.14 The number of herringbone packings strongly depends on the substitution of the carboxylates; especially, the p-substituent on the aromatic group of the carboxylates partially prevents the formation of the herringbone packing.
Thus, the crystallographic analyses of these less-soluble diastereomeric salts indicate that the dominant interactions in the crystals are 1) hydrogen-bonding interactions, which construct a supramolecular column, and 2) CH⋯π interactions, which pack the inter-columns tightly with each other through herringbone packings. The less-soluble diastereomeric salts were efficiently stabilised by these two kinds of interactions.
Effect of the α-alkyl group on the 2-arylalkanoic acids
With larger α-alkyl groups on the acid, the hydrogen-bond pattern in the less-soluble diastereomeric salts changed slightly from that shown in Fig. 4, although a columnar network was still formed around the 21-axis. This is thought to be caused by a steric repulsion between the alkyl group and 2·H+, since the alkyl group is in close contact with Ar(2) as shown in Fig. 3; a more serious repulsion would make the crystal unstable. The crystal structures and a schematic representation of the hydrogen-bond network in the less-soluble diastereomeric salts, (1S,2R)-2·(S)-6 and (1S,2R)-2·(S)-7, are shown in Fig. 5. For each case, a symmetrically independent 2·H+ molecule forms four hydrogen bonds with two carboxylate molecules and one 2·H+ molecule; the number of the hydrogen-bonded carboxylate molecules is different from that shown in Fig. 4. One 2·H+ molecule and one carboxylate molecule form a pair by two hydrogen bonds between the carboxylate oxygen and the ammonium hydrogen, and between the carboxylate oxygen and the hydroxy hydrogen of 2·H+ (Fig. 5, A and D). The other ammonium hydrogens of 2·H+ form hydrogen bonds with other pairs (Fig. 5, B and C). As a result, an infinite columnar hydrogen-bond network is formed. | ||
| Fig. 5 Crystal structures of less-soluble a) (1S,2R)-2·(S)-6 and b) (1S,2R)-2·(S)-7. Left, viewed down the b-axis; middle, columnar hydrogen-bond network; right, schematic representation of the hydrogen-bond network. The hydrogen atoms are omitted for clarity. The dotted lines and arrows show the hydrogen bonds and herringbone packings, respectively. The solid square represents the unit cell. | ||
Stacking of the aromatic rings is also achieved, as was observed in Fig. 3. However, the relative position of the aromatic and α-alkyl groups of the carboxylate in the hydrogen-bond network is different for both (1S,2R)-2·(S)-6 and (1S,2R)-2·(S)-7; this change of orientation accompanies a change in the absolute configuration of the carboxylate in the crystals. In these crystals, herringbone packing is achieved between adjacent Ar(2) and Ar(acid) within a single columnar hydrogen-bond network (indicated by arrows in Fig. 5). In addition, the inter-columns are also stabilised by herringbone packing.
In both (1S,2R)-2·(S)-6 and (1S,2R)-2·(S)-7, the most bulky substituent tends to avoid Ar(2). So the bulkiness of the α-alkyl group of the carboxylate strongly affects the structure of the hydrogen-bond network and the absolute configuration of the carboxylate in the less-soluble diastereomeric salts.
Crystal structure of the more-soluble diastereomeric salt and difference in stability between the less- and more-soluble diastereomeric salts
Single crystals of more-soluble diastereomeric salts are generally quite difficult to grow. However, we could determine the crystal structure of more-soluble (1S,2R)-2·(S)-4a (Fig. 6). In the crystal, there is also a columnar supramolecular hydrogen-bond network formed between the carboxylates and the ammoniums (Fig. 6b), as was found in the corresponding less-soluble diastereomeric salt. However, in more-soluble (1S,2R)-2·(S)-4a, the distance between the hydroxy group of 2·H+ and the carboxylate oxygen (hydrogen bond D) is significantly longer (ca. 0.2 Å) than that of less-soluble (1S,2R)-2·(R)-4a. Moreover, an IR study15 on the more- and less-soluble diastereomeric salts revealed that the absorption band for the hydroxy vibration of the more-soluble diastereomeric salt is shifted ca. 50 cm−1 upward from that of the less-soluble one. The shift of the infrared absorption band for the hydroxy vibration was also found for the combinations of (1S,2R)-2·(S)-3d/(1S,2R)-2·(R)-3d (110 cm−1) and (1S,2R)-2·(S)-4b/(1S,2R)-2·(R)-4b (60 cm−1). These results strongly suggest that the less-soluble diastereomeric salts are commonly stabilised by hydrogen bonds with the hydroxy group of 2·H+ much more than are the corresponding more-soluble diastereomeric salts. | ||
| Fig. 6 Crystal structure of more-soluble (1S,2R)-2·(S)-4a. a) Viewed down the b-axis (left) and columnar hydrogen-bond network (right) formed in (1S,2R)-2·(S)-4a. b) Schematic representation of the hydrogen-bond network. The dotted lines show hydrogen bonds. The hydrogen atoms are omitted for clarity. The dotted lines and arrows show the hydrogen bonds and herringbone packings, respectively. The solid square represents the unit cell. | ||
While there is some consistency in the pattern of the hydrogen-bond network in the less- and more-soluble diastereomeric salts, the characteristics of the packing of the aromatic groups are considerably different between less-soluble (1S,2R)-2·(R)-4a and more-soluble (1S,2R)-2·(S)-4a; in more-soluble (1S,2R)-2·(S)-4a, the most bulky phenyl group, Ar(4a), is close to Ar(2). Although this orientation would bring about steric repulsion between the aromatic rings, it allows intra-columnar herringbone packing between Ar(2) and Ar(4a). The different orientation of Ar(4a) against Ar(2) in the columnar hydrogen-bond network causes a difference in the number of effective herringbone packings between the less- and more-soluble diastereomeric salts. In less-soluble (1S,2R)-2·(R)-4a, three kinds of inter-columnar herringbone packings are achieved between Ar(2) and Ar(4a), as shown in Fig. 3,13 while only two (inter-columnar and intra-columnar) are achieved in more-soluble (1S,2R)-2·(S)-4a.
Thus, the comparison of the crystal structures of the less-soluble (1S,2R)-2·(R)-4a with that of the more-soluble (1S,2R)-2·(S)-4a reveals that the difference arises from hydrogen-bonding and CH⋯π interactions. The absolute conformation of (R)-4a favors such interactions, while that of (S)-4a does not.
The cell parameters of less-soluble (1R,2S)-2·(S)-3f and more-soluble (1S,2R)-2·(S)-3f are similar to each other, and even to those listed in Table 2,8 implying that similar networks are formed in these salts. This suggests that the chiral discrimination of the enantiomers of 2-arylalkanoic acids by enantiopure 2 would commonly occur during the formation and packing of columnar supramolecular hydrogen-bond networks.
Effect of the fixed cis-conformation of the amino alcohol
The dominant interactions in the salts of 2 with 2-arylalkanoic acids are hydrogen-bonding and CH⋯π interactions. Therefore, the elucidation of the geometrical relationship between the aromatic group(s) and the hydrogen-bond-forming groups in the amino alcohols 1 and 2 is important, in order to understand the difference in the structure of the columnar hydrogen-bond network between the salts of 2 and those of 1.Fig. 7 shows schematic drawings of the stackings of 1·H+ and 2·H+ along with the hydrogen-bond column. A comparison of the packing features of 2·H+ with those of 1·H+ shows that the direction of the C–N bond toward the columnar axis is not much different, and that the stacking of the aromatic groups is achieved in both hydrogen-bond columns. These facts strongly suggest that the hydrogen bonds formed by the ammonium group and the stacking of the aromatic rings play a principal role in the formation of the columnar network. A clear difference is found in the position of the hydroxy group. In the case of 1·H+, the hydroxy group is gauche to the phenyl group at C2, and the C1–O bond is parallel to the columnar axis. As a result, the hydroxy group is located apart from the hydrogen-bond column, and can participate in the hydrogen-bond network only from the outside. This geometry alters the role of the hydroxy group in the formation of the hydrogen-bond column in the presence or absence of a water molecule. In the case of 2·H+, the hydroxy group is anti to the aromatic group, and the C2–O bond is rather perpendicular to the columnar axis and takes almost the same direction as the C1–N bond against the columnar axis. This geometry allows the hydroxy group to penetrate deep into the hydrogen-bond column and would lead to the formation of a common and consistent hydrogen-bond network for the series of crystals in the present study.
 | ||
| Fig. 7 Schematic representations of the stackings of a) 1·H+ and b) 2·H+ along the columnar axis. The hydrogen atoms are omitted. The cylinder represents a hydrogen-bond network formed by the carboxylate oxygens and the ammonium hydrogens. | ||
Thus, the formation of a consistent columnar supramolecular hydrogen-bond network in the less- and more-soluble diastereomeric salts of enantiopure 2 with 2-arylalkanoic acids is thought to arise, not only from the fixed cis-conformation of the amino and hydroxy groups of 2, but also from the geometrical relationship between the aromatic and hydroxy groups.
Conclusions
The present results reveal that the discrimination of the enantiomers of 2-arylalkanoic acids by enantiopure 2 upon crystallisation was achieved via the formation of a consistent columnar supramolecular hydrogen-bond network. The cis-conformation of 2 results in an increase in resolving efficiency for racemic 2-arylalkanoic acids. In chiral discrimination, hydrogen-bonding interaction is found to be the most important factor. In addition, CH⋯π interaction is also suggested to play a role. The differentiation in stability between the less- and more-soluble diastereomer is thought to arise from a difference in the strength of these two kinds of interactions.Moreover, the relative orientation of the ammonium, hydroxy, and aromatic groups of the resolving agent, which induce significant interactions for stabilisation of the crystals, is found to be quite important; a comparison of the molecular structures of crystalline 1·H+ and 2·H+ indicates that the ammonium and aromatic groups play a principal role in the formation of the columnar network, while the role of the hydroxy group changes depending on its orientation. These results suggest that to design a resolving agent, one should pay particular attention to the kind, number, and orientation of such interacting groups. In other words, the present results demonstrate the possibility that the design of a resolving agent could be achieved by considering the combination and orientation of supramolecular synthons16 in a resolving agent.
Experimental
General
The infrared spectra were recorded on a Jasco IR-810 spectrophotometer, and the 1H and 13C NMR spectra were measured on a JEOL PMX-60SI or Varian Mercury-300 instrument using tetramethylsilane as an internal standard. Analytical HPLC was performed using Daicel Chiralcel OJ, OB, OD (eluent: hexane–propan-2-ol), or OJ-R (eluent: 0.5 M H3PO4–KH2PO4 buffer (pH 2)/CH3CN) columns with the detector wavelength at 254 nm. GC was performed using a Chrompak CHIRALDEX GT-A column with a helium carrier gas. The chromatograms were recorded on a HITACHI D02500 chromato-integrator.(1S,2R)-2, 3a, 4a, and 7 are commercially available. The preparations of other racemic acids were carried out as described in our previous paper.3 All solvents were distilled before use.
Resolution of 2-arylalkanoic acids with (1S,2R)-2
Equimolar amounts of racemic 2-arylalkanoic acid ( 3–7) (1–3 mmol) and (1S,2R)-2 (1–3 mmol) were placed in a 5-10 cm3 sample tube and suspended in a solvent (see Table 1). The sample tube was heated on a hot-plate until the suspension turned to a clear solution. The heating was continued for an additional 10 min, and then the sample tube was moved to a water bath kept at 30 °C and left standing for 12 h. During the standing, equilibrium between the crystals and the solution was achieved to afford reproducible results. The crystals which precipitated were collected by filtration and dried in vacuo for a few hours. After the crystals had been treated with 1 M hydrochloric acid, the liberated acid was extracted with ether (10 cm3), and the ethereal solution was dried over magnesium sulfate. Removal of the solvent in vacuo gave the acid, the enantiomeric excess of which was determined by HPLC or GC analysis. If necessary, the acid was converted to the corresponding methyl ester by treatment with diazomethane before the analysis.Preparation of single crystals
Single crystals of (1S,2R)-2·(R)-3a, (1S,2R)-2·(R)-3c, (1S,2R)-2·(R)-3e·H2O, (1S,2R)-2·(R)-4a, (1S,2R)-2·(R)-4b, (1S,2R)-2·(R)-6, and (1S,2R)-2·(R)-7 were directly prepared during the resolution experiments, as is indicated above.For (1R,2S)-2·(S)-3f, (1S,2R)-2·(S)-3f, and (1S,2R)-2·(S)-4a, an aqueous ethanol (water–ethanol = 5∶1, ca. 2 mL) solution of an equimolar mixture of the corresponding enantiopure acid (0.1 mmol) and amine (0.1 mmol) was placed in a sample tube (5 cm3), and the sample tube was left at 25 °C for two–seven days; the solvent was slowly evaporated to afford single crystals.
For (1R,2S)-2·(S)-3f and (1S,2R)-2·(S)-3f, only cell parameters could be determined. Mps and IR spectra for the above compounds are given in Table 3.
| Salt | Mp/°C | νmax/cm−1 |
|---|---|---|
| (1S,2R)-2·(R)-3a | 169.0–171.0 | 3450, 2970, 2100, 2000, 1610, 1540, 1395 |
| (1S,2R)-2·(R)-3c | 164.0–165.0 | 3240, 2050, 1610, 1550, 1395 |
| (1R,2S)-2·(S)-3e·H2O | 133.5–134.0 | 3600, 3500, 3280, 1610, 1550, 1515, 1390 |
| (1R,2S)-2·(S)-3f | 177.0–179.0 | 3250, 2090, 1600, 1540, 1390 |
| (1S,2R)-2·(S)-3f | 169.0–172.0 | 3450, 2120, 1600, 1490, 1390 |
| (1S,2R)-2·(R)-4a | 159.0–161.0 | 3200, 2970, 2700, 1520, 1400 |
| (1S,2R)-2·(S)-4a | 137.5–138.0 | 3250, 2970, 2940, 2880, 2100, 1540, 1400 |
| (1S,2R)-2·(R)-4b | 161.0–163.5 | 3200, 2120, 1610, 1525, 1390 |
| (1S,2R)-2·(S)-4b | 108.0–112.0 | 3260, 2100, 1610, 1550, 1530, 1390 |
| (1S,2R)-2·(S)-6 | 167.0–168.0 | 3430, 2970, 2000, 1620, 1560, 1400 |
| (1S,2R)-2·(S)-7 | 197.0–198.0 | 3050, 2940, 2850, 1605, 1590, 1490, 1380 |
Crystal-structure determination and refinement
The X-ray intensities were measured up to 2θ = 130° with graphite-monochromated Cu-Kα radiation (λ = 1.5418 Å) on a Mac Science MXC18 four-circle diffractometer by a 2θ-ω scan. All of the data were collected at 293 K. The cell dimensions were obtained by least-squares analyses of the setting angles of 20 reflections (50 < 2θ < 60°). The intensities and orientation of the crystals were checked by three standard reflections every 100 reflections.The structures were solved and refined by applying the CRYSTAN-GM package;17 the direct method (SIR9218) followed by normal heavy-atom procedures, and full-matrix least-squares refinement with all non-hydrogen atoms anisotropic and hydrogens in calculated positions with thermal parameters equal to those of the atom to which they were bonded. Atomic coordinates, thermal parameters, bond lengths and angles for all diastereomeric salts have been deposited at the Cambridge Crystallographic Data Centre. CCDC reference number 188/194. See http://www.rsc.org/suppdata/p2/a9/a905566e for crystallographic files in .cif format.
Acknowledgements
The present work was supported by Grants-in-Aid for Scientific Research (Nos. 09450330 and 10750618) from the Ministry of Education, Science, Sports and Culture of Japan and by Nagase Science and Technology Foundation.References
- J. Jacques , A. Collet and S. H. Wilen , Enantiomers, Racemates, and Resolutions; Krieger Publishing Company, Malabar, Florida, 1994. Search PubMed.
- For examples of novel resolving agents, see: A. D. van der Haest, H. Wynberg, F. J. J. Leusen and A. Bruggink, Recl. Trav. Chim. Pays-Bas, 1993, 112, 230 Search PubMed; D. Kozma, M. Ács and E. Fogassy, Tetrahedron, 1994, 50, 6907 Search PubMed; B. Eric and M. Moudachirou, Tetrahedron, 1994, 50, 10309 CrossRef CAS; M. Pallavicini, E. Valoti, L. Villa and O. Piccolo, Tetrahedron: Asymmetry, 1996, 7, 1117 CrossRef.
- K. Kinbara, Y. Kobayashi and K. Saigo, J. Chem. Soc., Perkin Trans. 2, 1998, 1767 RSC.
- T. Manimaran and A. A. Potter , US Patent 5,162,576, 1992. Search PubMed.
- More precisely, the optimisation of the resolution conditions can be done on the basis of binary and ternary phase diagrams.6,7 In the present study, instead of optimisation, we adjusted the quantity and ratio of the mixed solvent so that the resolving efficiency was as high as possible..
- E. Fogassy, F. Faigl and M. Ács, Tetrahedron, 1985, 41, 2837 CrossRef CAS; F. J. J. Leusen, J. H. Noordik and H. R. Karfunkel, Tetrahedron, 1985, 49, 5377 CrossRef CAS; D. Kozma, G. Pokol and M. Ács, J. Chem. Soc., Perkin Trans. 2, 1992, 435 RSC; E. Collet , in Comprehensive Supramolecular Chemistry, D. N. Reinhoudt, ed., Pergamon, Oxford, 1996, vol. 1, pp. 113–149; Search PubMed; E. J. Ebbers, B. J. M. Plum, G. J. A. Ariaans, B. Kaptein, Q. B. Broxterman, A. Bruggink and B. Zwanenburg, Tetrahedron: Asymmetry, 1997, 8, 4047 Search PubMed; E. Ebbers, G. J. A. Ariaans, B. Zwanenburg and A. Bruggink, Tetrahedron: Asymmetry, 1998, 9, 2745 CrossRef CAS.
- J. Viret, H. Patzelt and A. Collet, Tetrahedron Lett., 1986, 27, 5865 CrossRef CAS; T. Shiraiwa, Y. Sado, S. Fujii, M. Nakamura and H. Kurokawa, Bull. Chem. Soc. Jpn., 1987, 60, 824 CAS.
- In addition to the salts in Table 2, the cell parameters and the space group could be determined for (1R,2S)-2·(S)-3f and (1S,2R)-2·(S)-3f (more soluble). (1R,2S)-2·(S)-3f: P21, a = 14.275(7) Å, b = 6.203(3) Å, c = 10.736(6) Å, β = 106.80 (4)°, V = 910.0(8) Å3. (1S,2R)-2·(S)-3f: P21, a = 14.620(9) Å, b = 6.156(5) Å, c = 10.261(5) Å, β = 99.08 (5)°, V = 912(1) Å3..
- K. Kinbara, Y. Hashimoto, Y. Sukegawa, H. Nohira and K. Saigo, J. Am. Chem. Soc., 1996, 118, 3441 CrossRef CAS.
- A table of N⋯O and O⋯O distances in the diastereomeric salts of (1S,2R)-2 with 3–7 is deposited as supporting information (Table S1)..
- Concerning the favorable location of hydrogens around hydrogen acceptors, see: R. Taylor and O. Kennard, Acc. Chem. Res., 1984, 17, 320 Search PubMed; J. P. Glusker, Mol. Cryst. Liq. Cryst., 1992, 211, 75 CrossRef CAS.
- A. Gavezzotti and G. R. Desiraju, Acta. Crystallogr., Sect. B, 1988, 44, 427 CrossRef; G. R. Desiraju and A. Gavezzotti, Acta. Crystallogr., Sect. B, 1989, 45, 473 CrossRef.
- A table of the angles and lengths between the stacking aromatic rings is deposited as supporting information (Table S2 and Table S3)..
- M. Nishio
, M. Hirota
and Y. Umezawa
, CH⋯π Interaction: Evidence, Nature, and Consequences, Wiley-VCH Publication: Weinheim, Berlin, 1998.
Search PubMed.
- L. J. Bellamy and R. J. Pace, Spectrochim. Acta, 1969, 25, 319 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- CRYSTAN GM, A Computer Program for the Solution and Refinement of Crystal Structures for X-ray Diffraction Data (MAC Science Corporation). Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
Footnote |
| † Data relating to the salts of (1S,2R)-2 with 3–7 are available as supplementary data. For direct electronic access see http://www.rsc.org/suppdata/p2/a9/a905566e, otherwise available from BLDSC (SUPPL. NO. 57677, 4 pp.) or the RSC Library. See Instructions for Authors available via the RSC web page (http://www.rsc.org/authors). |
| This journal is © The Royal Society of Chemistry 2000 |