Reduced corannulenes: 1,8-dicorannulenyloctane anions, a supramolecular octaanion
Elad Shabtaia, Roy E. Hoffmana, Pei-Chao Chengb, Eric Bayrdb, Dorin V. Predab, Lawrence T. Scott*b and Mordecai Rabinovitz*a
aDepartment of Organic Chemistry, The Hebrew University of Jerusalem, Givat Ram, Jerusalem, Israel 91904
bDepartment of Chemistry, Boston College, Eugene F. Merkert Chemistry Center, Chestnut Hill, MA 02467-3860, USA
First published on UnassignedUnassigned23rd December 1999
Abstract
Two corannulenes joined by an octamethylene chain as in 1,8-dicorannulenyloctane (3) can be reduced with lithium, sodium, potassium or cesium in [D8]THF to give a purple tetraanion. In this state, each corannulene moiety behaves as an independent, strongly paratropic dianion that is best described as an aromatic, 6 π-electron, cyclopentadienyl anion in the core, suspended within an antiaromatic, 16 π-electron, [15]annulenyl anion around the rim. The bowl-to-bowl inversion barrier of the tethered corannulene dianions was determined by variable temperature 1H NMR spectroscopy to be 8.8 ± 0.3 kcal mol−1 with potassium counter ions and 9.2 ± 0.3 kcal mol−1 with cesium counter ions. It is the first time that the barrier for a bowl-shaped charged species was determined. As expected, charging reduces the energy barrier for bowl-to-bowl inversion relative to that in the neutral hydrocarbon (10.9 ± 0.3 kcal mol−1). Further reduction of the hydrocarbon with lithium, potassium or cesium gives an octaanion, but no further reduction could be achieved with an excess of sodium metal. The bowl-to-bowl inversion barrier of the octaanion is too low to be determined by variable temperature 1H NMR spectroscopy. Ion diffusion measurements and the failure of 3 to form mixed sandwich compounds with corannulene (1) when it is reduced in the presence of an excess of corannulene are consistent with intramolecular stacking (rather than intermolecular stacking) of the two tethered corannulene tetraanions to form lithium ion-bound sandwiches of the type seen previously for unsubstituted corannulene.
Introduction
The corannulene tetraanion (14−), being the first member of a new class of exceptionally stable, high-order sandwich compounds, has added a striking new feature to the landscape of organolithium chemistry. The bowl shaped corannulene molecule (1) is known to have a doubly degenerate low lying LUMO that can accept up to four extra electrons.1 We have previously reported![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 1 the properties of corannulene tetraanion (14−) and the unexpected aggregation of this species to form high order molecular sandwiches upon lithium reduction.2 Ion solvation equilibrium studies of 14− by 7Li NMR spectroscopy provided evidence for the presence of two kinds of Li cations at low temperatures (ratio 1∶1) which exchange environments rapidly on the NMR timescale at room temperature (ΔG*265 = 13.2 kcal mol−1). The 7Li chemical shifts reflect the location of four lithium cations sandwiched between the two layers of the dimeric hydrocarbon superstructure, with two more above and two more below the sandwich (2).2
1 the properties of corannulene tetraanion (14−) and the unexpected aggregation of this species to form high order molecular sandwiches upon lithium reduction.2 Ion solvation equilibrium studies of 14− by 7Li NMR spectroscopy provided evidence for the presence of two kinds of Li cations at low temperatures (ratio 1∶1) which exchange environments rapidly on the NMR timescale at room temperature (ΔG*265 = 13.2 kcal mol−1). The 7Li chemical shifts reflect the location of four lithium cations sandwiched between the two layers of the dimeric hydrocarbon superstructure, with two more above and two more below the sandwich (2).2Recently, we have succeeded in characterizing all five reduction states of corannulene with lithium and have suggested a full reduction pathway from the neutral compound to the dimer of the tetraanion.3 The extremely high field shift of the protons in the dianion (−5.6 ppm) is typical for molecules with paratropic ring current effects.4 The change in proton chemical shifts from 7.94 ppm in the neutral molecule to −5.6 ppm in the dianion and then back to 6.85 ppm in the tetraanion fits well the description of 1 as an “annulene within an annulene” originally suggested by Barth and Lawton.5
In view of the formation of the octalithio dimer of 14−i.e.2, it would be reasonable to expect two corannulene units tethered by a oligomethylene chain, e.g., 1,8-dicorannulenyloctane (3), to exist as an intramolecular sandwich in the charged form. In principle, a preorganized system such as 3, when reduced with alkali metals, could form either intra- or inter-molecular sandwiches. In the case of intermolecular aggregation, the forced proximity of the two corannulene units might be expected to lead to charge repulsion between the two sandwiches and thereby to an increase in the energy of the entire system. In the case of intramolecular aggregation, on the other hand, a long enough tether should lead to stabilization of the system.
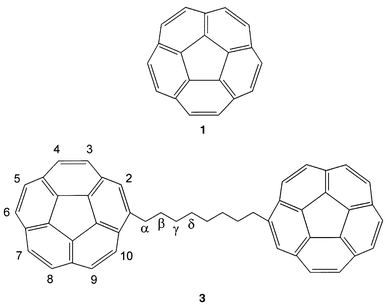
Using the dimethylmethanol derivative of corannulene (1),6 Scott etal. were able to follow the dynamic behavior of the polycyclic system and thereby determine the bowl-to-bowl inversion barrier of corannulene (ΔG*209 = 10.2 ± 0.2 kcal mol−1). In the case of 3, each methylene unit of the tether can in principle serve as a chiral probe. The complications7 that render an isopropyl substituent unsuitable as a chiral probe on the charged derivatives of 1 can be overcome by observing the signals from a remote methylene group in 3, one that is far away from the “battle field” of charge and magnetic anisotropy. Using a remote methylene group in 3 that is far enough removed from the effect of the induced magnetic field of the charged moieties, yet still sensitive to the bowl-to-bowl inversion process, one should be able to follow the dynamic process and determine, for the first time, the bowl-to-bowl inversion barrier of the charged species.

Herein we report the formation of tetra- and octa-anions derived from 1,8-dicorannulenyloctane (3) with lithium, sodium, potassium and cesium and their 1H and 7Li NMR spectra. The bowl-to-bowl inversion barriers of the corannulene moieties in the neutral form and in the tetraanion of 3 with potassium and cesium cations have also been measured. The quenched products of the anions derived from 1, i.e.4 and 5, and the reduction of 1 with five alkali metals (Li, Na, K, Rb and Cs) are also described.
Quenching products of corannulene anions
The NMR data of corannulene tetraanion (14−) show a diamagnetic ring current effect where an excess of electron density resides on the rim, with only a relatively small amount on the hub.1 This supports the model of an “annulene within an annulene”.5a Molecular orbital calculations, however, suggested that the tetraanionic structure may be more complicated than the highly symmetrical “anion within a trianion”.1a Indirect information on the π-electron distribution in anions can be achieved from the structure of the quenching products. The quenching products (D2O) of the di- and tetra-anion of corannulene were determined to have molecular masses of m/z 252 and 254, respectively.1 However, the structures of these two molecules were not previously studied.The reduction of corannulene (1) with lithium to its dianion and tetraanion was validated by the detection of the known NMR spectra. The quenching reaction with water under an inert atmosphere led to the formation of dihydrocorannulene (4)8 and tetrahydrocorannulene (5), respectively, as the dominant products. The structures of both products were determined by complete 2-D NMR analyses. As a result of fast electron transfer, both quenching experiments yield tetrahydrocorannulene, dihydrocorannulene and corannulene. According to the “annulene model” the strongly paramagnetic dianion is best described as an aromatic, 6π-electron, cyclopentadienyl anion in the core, suspended within an antiaromatic, 16π-electron, [15]annulenyl anion. However, the NMR spectrum of dihydrocorannulene (4) shows that the two added protons become attached to the same six-membered ring in a way that destroys the aromaticity of only one of the five benzene rings (Fig. 1). Similar behavior was found in the quenching product of the tetraanion (5). Where the calculation points to a 6π-electron, cyclopentadienyl anion in the core, suspended within an aromatic, 18π-electron, [15]annulenyl anion around the rim, the four added protons in the quenching product of the tetraanion become attached to vicinal carbons on the perimeter of the system. It seems that the quenching products reflect more the tendency to higher stability (thermodynamic products) rather than the charge distribution of the anion (kinetic products).
 | ||
| Fig. 1 The quenched products of the anions derived from corannulene. | ||
Reduction of corannulene with alkali metals
From the early study of corannulene anions, the highest reduction stage of corannulene (tetraanion) could be achieved only with lithium. Reduction with sodium and potassium led only to the dianion. The explanation for this behavioral difference was based mainly on the differences in ion pairing equilibria and the tendency of the lithium cations to aggregate and thus to stabilize the anions. Recently, we succeeded in achieving the higher reduction stage of corannulene also with higher atomic number alkali metals.Reduction of corannulene (1) with all alkali metals under gentle conditions led to the appropriate dianions. The observation of only small differences in the UV-Vis spectra of the dianion solutions (up to 4 nm) indicated loosely bonded interaction of the cations, as was previously assumed. Further reduction with lithium, potassium, rubidium and cesium gave the highly charged tetraanions. However, with sodium as the reducing metal, no further reduction could be achieved beyond the dianion. In contrast to the highly stable tetraanion with lithium, the tetraanions with potassium, rubidium, and cesium could be detected only after a long period of time and showed very broad signals. The spectra of these tetraanions could be obtained only at a low temperature, indicating an equilibrium with a paramagnetic species (trianion radical) or an energetically close triplet state. The NMR chemical shifts are summarized in Table 1. The similarity of the reduced species with potassium, rubidium, and cesium, and the difference of these species from the lithium reduction product, point to the existence of different types of solvation states. In contrast to earlier experiments with lithium metal, reduction of a mixture of corannulene (1) and tert-butylcorannulene with cesium did not lead to the appearance of a mixed dimer. Thus, the larger alkali metals probably form monomeric tetraanions.
| Tetraanion | Dianion | |||
|---|---|---|---|---|
| Carbon | Proton | Carbon | Proton | Metal |
| 86.6 | 120 | |||
| 95.0 | 6.85 | 154 | −5.60 | Li |
| 112.3 | 203 | |||
| 117.9 | ||||
| 154.5 | −5.10 | Na | ||
| 200.6 | ||||
| 95.6 | 119.1 | |||
| 102.5 | 6.51 | 153.5 | −5.58 | K |
| 117.8 | 204.7 | |||
| 95.7 | 119.0 | |||
| 103.8 | 6.54 | 153.4 | −5.65 | Rb |
| 118.8 | 206.4 | |||
| 98.7 | 118.9 | |||
| 106.2 | 6.50 | 153.8 | −5.60 | Cs |
| 121.0 | 207.3 | |||
Reduction of 1,8-dicorannulenyloctane to its tetraanion and octaanion
Tethered corannulene 3 was prepared from corannulene by Friedel–Crafts acylation with suberoyl chloride, followed by Clemmensen reduction. Controlled reduction of 3 with lithium in [D8]THF solution under high vacuum gave color changes similar to those observed with corannulene: green, purple and finally a brownish red solution. This corresponds to three stages of charging: radical anion, dianion and tetraanion of each corannulene unit.3In contrast to the single line in the 1H NMR spectrum of corannulene (1) at δH 7.94,1 the decrease in symmetry in 3 gives rise to eight doublets (δH 7.74, 7.78, 7.80, 7.80, 7.81, 7.81, 7.84 and 7.98) and one singlet (δH 7.61) from the ring protons. The four multiplets (δH 1.49, 1.59, 1.94 and 3.17) were assigned to protons of the four different sets of methylene groups in the bridge (Table 1).
The 1H NMR spectrum of the tetraanion of 3 with lithium as counter ion ([D8]THF, T = 200 K) consists of one broad peak for the corannulene protons (δH −3.5 to −4.7) and three peaks due to protons on six of the methylene units at a relatively high field for alkane protons (δH 0.28, −0.37 and −1.94). This system represents two paratropic doubly charged corannulene moieties linked by a polymethylene chain. The chemical shift of the benzylic protons upon charging is influenced by the induced paramagnetic “ring current,” in addition to its proximity to the charged core. The signal for these methylene protons, which is covered by the broad signal from the protons on the corannulene dianion moiety at room temperature, appears only at very low temperature (T < 175 K), where the broad peak from the ring protons is narrowed. An extremely high-field chemical shift of δ −3.3 is then observed for the benzylic methylene protons.
Further reduction of 3 yields a new diamagnetic charged product: the octaanion. The 1H NMR spectrum of the octaanion with lithium ([D8]THF, T = 200 K) consists of four broad peaks in the range of δH 2.2 to 3.2 due to the protons on the tether and another set of peaks between δH 6.7 and 7.2, in the same region as the signal (δH 6.85) of the corannulene tetraanion (14−),1,2 which were assigned to the protons of the corannulene moieties. Quenching the octaanion with dry oxygen regenerates the neutral compound (3) in quantitative yield, thus discounting the possibility that the new NMR signals might be the result of some more elaborate chemical reaction.
The 7Li NMR spectrum of 38− shows two singlets (δLi
−4.3 and −11.5) which coalesce at 275 K to give one sharp line with a chemical shift of δLi
−8.3. The behavior of the 7Li spectrum is very similar to that of the corannulene tetraanion dimer (2), where the two singlets in the 7Li NMR spectrum (δLi
−4.5 and −11.7) coalesce at 265 K into a single peak at δLi
−8.1.2 The barriers for the Li+ exchange process in systems 2 and 38− are similar (ΔG*265 = 13.2 and ΔG*275 = 13.7 kcal mol−1 for 14− and 38−, respectively).
1,8-Dicorannulenyloctane (3) was also reduced with potassium and cesium to the tetra- and octa-anions, which were characterized by their 1H NMR spectra. Except for slight differences in the chemical shifts, the behavior of 3 under reduction with these alkali metals is similar to that described above with lithium. Reduction with sodium, on the other hand, led only to the tetraanion. The 1H NMR chemical shifts are summarized in Table 2.
| Compound | δ | γ | β | α | Protons 2–10c | |
|---|---|---|---|---|---|---|
| a At room temperature. b At 200 K. c The full assignment of 3 is: δH 7.61 [s, 2H, H-2], 7.74 [d, JHH = 8.6 Hz, 2H, H-3], 7.78 [d, JHH = 8.6 Hz, 2H, H-4], 7.80 [AB system, 4H, H-5, H-6], 7.81 [AB system, 4H, H-7, H-8], 7.84 [d, JHH = 8.6 Hz, 2H, H-9], 7.98 [d, JHH = 8.6 Hz, 2H, H-10]. d Observable only at T ≤ 175 K. | ||||||
| 3 | 1.49 | 1.59 | 1.94 | 3.17 | 7.61–7.98 | |
| 34− | 0.28 | −0.37 | −1.94 | −3.3d | −3.5 to −4.7 | Li |
| 0.49 | −0.14 | −1.60 | −2.5 to −4.7 | Na | ||
| −0.30 | −0.98 | −2.80 | −3.3 to −4.7 | K | ||
| −0.27 | −0.94 | −2.71 | −3.3 to −4.7 | Cs | ||
| 38− | 2.2–3.2 | 6.7–7.2 | Li | |||
| 1.96 | 2.18 | 2.66 | 3.05 | 6.2–6.8 | K | |
| 1.89 | 2.00 | 2.24 | 2.94 | 6.0–6.6 | Cs | |
Bowl-to-bowl inversion measurements
Variable temperature 1H NMR spectroscopy was used to study the bowl-to-bowl inversion processes. The 1H NMR spectrum of neutral 3 at room temperature shows four types of methylene groups. Decreasing the temperature to 170 K causes a broadening of three of the signals, while the triplet from the benzylic group is split into two triplets. The coalescence temperature for these two benzylic signals is 230 K, and the barrier for bowl-to-bowl inversion is therefrom found to be ΔG*230 = 10.9 ± 0.3 kcal mol−1. This barrier height matches closely the ΔG*209 = 10.2 ± 0.2 kcal mol−1 found previously for corannulene with a dimethylmethanol substituent.6The 1H NMR spectra of 34− with all four metals (lithium, sodium, potassium, and cesium) show three signals assigned to twelve of the tether methylene protons, while the signals from the benzylic protons are covered by the rather broad signal from the corannulene protons. Decreasing the temperature shows an effect of the counter cation on the appearance of the signals from the methylene protons. Whereas the 34− obtained by reduction with lithium and sodium still gives only broad lines for the interior methylene groups (β, γ and δ) at 170 K, the 34− obtained by reduction with potassium or cesium gives a split signal for the protons of the γ methylene group at this temperature. Following the dynamic process in both the potassium and cesium tetraanions enabled us to measure barriers of ΔG*186 = 8.8 ± 0.3 and ΔG*196 = 9.2 ± 0.3 kcal mol−1, respectively. These low barriers are assumed to be associated with bowl-to-bowl inversion and are in remarkable agreement with Rabideau’s calculations (ΔG* = 7.9–9.2 kcal mol−1).9 The reduction in barrier height relative to that for the neutral system can be rationalized as a consequence of the need for better conjugation, and thus greater planarity, of the highly charged system.
For all the octaanions derived from 3 the four signals arising from the methylene groups can be observed in the 1H NMR only at T > 230 K. Decreasing the temperature causes line broadening and finally disappearance of the signals of all the salts (Li, K and Cs), thus preventing quantitative measurement of dynamic processes. It has previously been shown2 that the dimers of monosubstituted corannulene tetraanions undergo bowl-to-bowl inversion with a very low barrier (<5 kcal), and the same appears to be true for 38−.
Intramolecular vs. intermolecular stacking of the octaanion
From an examination of the calculated geometry of the octalithio dimer of 14−i.e.2, we believe that a tether of eight carbon atoms should be long enough (≈8 Å) for the two corannulene units in 38− to form either intra- or inter-molecular ion-bound sandwiches. In the absence of other data, however, the 1H NMR spectra of 38− do not allow an unambiguous assignment to be made, since only small differences in chemical shifts would be expected for the two charged forms. We therefore attempted to generate “mixed sandwiches” in which one or both of the reduced corannulene moieties in 38− would be stacked with 14−. Such mixed sandwiches have previously been seen spectroscopically in the co-reduction of corannulene and tert-butylcorannulene.2 In the present case, reduction of a mixture of 1 and 3 (10∶1) in [D8]THF yielded only the well characterized 2, while the signals for 38− could hardly be seen due to low concentration, and no new signals for a mixed sandwich between 38− and 14− appeared. This observation argues in favor of intramolecular sandwich formation in 38−.Another way to gain some indication of the aggregation state of 38− is to compare its “size” with that of 2, the dimer of 14−. In the case of an intermolecular interaction, its size should be more than twice that of the dimer of 14−, and the ratio between the sizes of the charged and neutral forms would be similar for 3 and 1. In the case of an intramolecular interaction, on the other hand, we would expect the size of 38− to be only slightly larger that of the dimer of 14− and the ratio between the charged and the neutral forms of 3 to be smaller than that of 1.
The Stokes–Einstein relation (D = kBT/6πηr), which correlates the size of a particle with its diffusion coefficient in solution, can be used to get an indication of the particle size. This correlation applies accurately, however, only for spherical particles that are much larger than the solvent molecules.10 No simple formula exists for molecules that are not spherical, as in the cases of 1 and 3 and their anions. Even so, preliminary diffusion measurements of polycyclic anions11 have shown that there is a strong dependence of the diffusion coefficient on the degree of solvation. Thus, although we cannot derive the absolute sizes of the unsolvated species from their diffusion coefficients, the similar degrees of solvation expected for 1 and 3, and for their anions, should allow comparisons of their relative diffusion coefficients to be used as an indication of relative particle sizes.
Diffusion coefficients were measured using the pulsed gradient stimulated echo (PGSTE) 1H NMR technique12,13 in [D8]THF solution at 298.5 ± 0.5 K.14 We have found a significant reduction in the diffusion constants upon charging, as has been reported previously for 1 and similar compounds.15 The fact that no significant changes were observed in the diffusion coefficient of the THF, which serves as an internal standard, proves that the observed changes in the diffusion coefficients are not due to a change in the sample viscosity following the reduction. Since hydrocarbon 3 bears an alkyl chain we expect it to be slightly larger than a hypothetical dimer of 1. The diffusion constant of 3 (8.51 × 10−10 m2 s−1) is 0.58 times that of 1 (1.45 × 10−9 m2 s−1), which translates to 0.78 times that of the hypothetical dimer of neutral 1.16 The diffusion constant of 38− (5.5 × 10−10 m2 s−1), by comparison, is 0.77 times that of the observed dimer of 14− (7.16 × 10−10 m2 s−1). These results are likewise consistent with intramolecular sandwich formation in 38− and are less compatible with intermolecular sandwich formation.
Conclusions
Bowl-to-bowl inversion barriers of corannulene dianions have been measured for the first time and found to be ca. 2 kcal mol−1 lower than that of the corresponding neutral corannulene. Tethering two corannulene units together by an octamethylene chain leads to formation of an intramolecular sandwich superstructure analogous to that of the corannulene tetraanion dimer 2. The 1H NMR and the dynamic 7Li NMR spectra of 38−/Li8 mimic those of the corannulene tetraanion dimer 2.Experimental
NMR analyses were performed on a Bruker DRX400 FT spectrometer operating at 400.13, 100.62 and 155.51 MHz for 1H, 13C and 7Li, respectively. Full structure assignments were obtained by applying 2-D NMR spectroscopic methods such as COSY and NOESY.The reduction process
Lithium metal was introduced as a wire (or distilled in the case of sodium, potassium, rubidium and cesium) into the upper part of an extended NMR tube containing the corannulene derivative (≈3 mg) then dry [D8]THF was vacuum transferred and the tube sealed under vacuum. The solution was brought into contact with the metal by repeatedly inverting the tube. Formation of the tetraanion and the octaanion was detected by 1H NMR spectroscopy.Quenching of the anions
All anions were quenched with oxygen or D2O. The oxygen quenching experiments were carried out by bubbling the gas into the NMR sample tube under anhydrous conditions, followed by the detection of the NMR spectrum. The quenching products with D2O were obtained by pouring the anion solution into D2O, under an inert atmosphere. After purification the products were fully characterized by 2-D NMR techniques.1,8-Dicorannulenyloctane-1,8-dione
Under a nitrogen atmosphere at 0 °C, 213 mg (1.6 mmol) of aluminum chloride were stirred with 10 mL of freshly distilled methylene chloride, and 0.11 mL (0.6 mmol) of suberoyl chloride was added. After 20 min, 100 mg (0.4 mmol) of corannulene were added to the mixture, and a yellow suspension formed immediately. After one hour of stirring at 0 °C, the reaction mixture was allowed to warm to room temperature and stirred at that temperature for 36 h. The reaction mixture was then washed with 3 × 30 mL of 10% hydrochloric acid and 3 × 30 mL of saturated aqueous sodium chloride. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated to dryness under reduced pressure. The crude product was purified by column chromatography (silica gel) using methylene chloride as eluent to give 48.0 mg (38%) of 1,8-dicorannulenyloctane-1,8-dione as a yellow solid, mp 187–188 °C. 1H NMR (400 MHz, CDCl3, 298K): δ 8.53 (d, J = 9.0 Hz, 2H), 8.46 (s, 2H), 7.85–7.77 (m, 14H), 3.21 (t, J = 7.3 Hz, 4H), 1.89 (m, 4H), 1.55 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 202.95, 138.08, 137.20, 137.17, 136.54, 135.95, 135.69, 132.66, 131.66, 131.52, 131.49, 129.51, 129.04, 128.97, 128.87 (2C), 128.24, 128.11, 127.81, 127.76 (2C), 40.98, 29.90, 25.36. IR (neat): 2930m, 2853w, 1669s, 1457m, 1435m, 832s cm−1. MS (FAB): m/z (relative intensity) 639 (25, M+ + H) 638 (10, M+), 613 (8), 460 (46), 341 (18), 307 (100), 291 (40). HRMS: Calc. for C48H31O2 (FAB, M+ + H) m/z 639.2324; Found 639.2326.1,8-Dicorannulenyloctane (3)
Three chunks (excess) of freshly prepared Zn(Hg) amalgam17 and 50 mg (0.08 mmol) of 1,8-dicorannulenyloctane-1,8-dione were stirred in a mixture of 3.5 mL of concentrated hydrochloric acid, 1.5 mL of distilled water, and 2.0 mL of toluene. The reaction mixture was heated to reflux (110 °C) for 12 hours and then allowed to cool to room temperature. Ten milliliters of distilled water were added, and the mixture was extracted repeatedly with methylene chloride. The combined organic extracts were washed with saturated aqueous sodium bicarbonate, dried over anhydrous magnesium sulfate, filtered, and concentrated to dryness under reduce pressure. The crude product was purified by preparative thin layer chromatography (silica gel) using 1∶1 hexane–methylene chloride as eluent to give 27 mg (56%) of 1,8-dicorannulenyloctane (3) as yellow crystals, mp 176–179 °C. 1H NMR (400 MHz, CDCl3, 298 K): δ 7.93 (d, J = 8.8 Hz, 2H), 7.82 (d, J = 8.8 Hz, 2H), 7.81–7.75 (m, 10H), 7.73 (d, J = 8.8 Hz, 2H), 7.57 (s, 2H), 3.15 (t, J = 7.6 Hz, 4H), 1.95 (m, 4H), 1.57 (m, 4H), 1.47 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 142.74, 136.66, 136.58, 136.53, 136.38, 135.43, 132.00, 131.55, 131.08 (3C), 127.71 (2C), 127.68, 127.59, 127.49 (2C), 127.27, 125.77, 125.59, 34.00, 33.02, 30.27, 30.22. HRMS: Calc. for C48H34 (M+) m/z 610.2660; Found 610.2656.1,2-Dihydrocorannulene (4)
1H NMR (400 MHz, CDCl3; 298 K): δ 7.72 [2H, d, J = 9.0 Hz, 6-H], 7.69 [2H, d, J = 9.0 Hz, 5-H], 7.65 [2H, d, JHH = 8.0 Hz, 4-H], 7.23 [2H, d, JHH = 8.0 Hz, 3-H], 3.38 [4H, s, 1-H]. 13C NMR (100 MHz, CDCl3): δ 127.9 [C-6], 127.5 [C-5], 126.9 [C-4], 126.3 [C-3], 27.3 [C-1].1,2,2a,3-Tetrahydrocorannulene (5)
1H NMR (400 MHz, CDCl3; 298 K): δ 7.61 [1H, d, JHH = 9.0 Hz, 7-H], 7.58 [1H, d, JHH = 9.0 Hz, 8-H], 7.55 [1H, d, JHH = 8.2 Hz, 6-H], 7.52 [1H, d, JHH = 8.2 Hz, 9-H], 7.10 [1H, d, JHH = 8.2 Hz, 5-H], 7.09 [1H, d, JHH = 8.2 Hz, 10-H], 5.78 [1H, m, 4-H], 4.51 [1H, m, 2a-H(ax)], 3.69 [1H, m, 3-H(ax)], 3.32 [1H, m, 1-H(ax)], 3.15 [1H, m, 3-H(eq)], 3.09 [1H, m, 2-H(ax)], 2.77 [1H, m, 1-H(eq)], 2.22 [1H, m, 2-H(eq)]. 13C NMR (100 MHz, CDCl3): δ 150.8 [C-4b], 148.0 [C-10b], 140.4 [C-6b], 139.9 [C-8b], 135.7 [C-2b], 134.2 [C-4a], 133.8 [C-10a], 129.9 [C-6a], 129.4 [C-8a], 126.0 [C-7], 125.9 [C-8], 125.4 [C-10], 125.1 [C-5], 125.0 [C-6], 125.0 [C-9], 121.8 [C-4], 44.8 [C-2a], 31.7 [C-3], 28.6 [C-2], 27.2 [C-1].Acknowledgements
We are grateful to the U.S.-Israel Binational Science Foundation and the U.S. Department of Energy for financial support for this work.References
- (a) A. Ayalon, M. Rabinovitz, P. C. Cheng and P. W. Rabideau, Angew. Chem., 1992, 104, 1691; Search PubMed; (b) A. Ayalon, M. Rabinovitz, P. C. Cheng and P. W. Rabideau, Angew. Chem., Int. Ed. Engl., 1992, 31, 1636. Search PubMed.
- A. Ayalon, A. Sygula, P. C. Cheng, M. Rabinovitz, P. W. Rabideau and L. T. Scott, Science, 1994, 265, 1065 CAS.
- M. Baumgarten, L. Gherghel, M. Wagner, A. Weitz, M. Rabinovitz, P. C. Cheng and L. T. Scott, J. Am. Chem. Soc., 1995, 117, 6254 CrossRef CAS.
- M. Rabinovitz, Top. Curr. Chem., 1988, 146, 99 Search PubMed; K. Müllen, T. Meul, P. Schade, H. Schmickler and E. Vogel, J. Am. Chem. Soc., 1987, 109, 4992 CrossRef.
- (a) W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1992, 1966, 88, 380; Search PubMed; (b) W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1971, 93, 1730. Search PubMed.
- L. T. Scott, M. M. Hashemi and M. S. Bratcher, J. Am. Chem. Soc., 1992, 114, 1920 CrossRef CAS.
- P. W. Rabideau,personal communication..
- A. Borchardt, A. Fuchicello, K. V. Kilway, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 1921 CrossRef CAS.
- A. Sygula and P. W. Rabideau, J. Mol. Struct. (THEOCHEM), 1995, 333, 215 CrossRef CAS.
- J. P. Hansen and I. R. McDonald , Theory of Simple Liquids, Academic Press, London, 1976. Search PubMed.
- R. E. Hoffman, E. Shabtai, M. Rabinovitz, V. S. Iyer, K. Müllen, A. K. Rai, E. Bayrd and L. T. Scott, J. Chem. Soc., Perkin Trans. 2, 1998, 1659 RSC.
- This experiment is also called pulsed magnetic field stimulated spin echo (PFG-SSE).
- J. E. Tanner, J. Phys. Chem., 1970, 52, 2523 CrossRef CAS; P. Stilbs, Prog. NMR Spectrosc., 1987, 19, 1 Search PubMed; R. M. Cotts, M. J. R. Hoch, T. Sun and J. T. Markert, J. Magn. Reson., 1989, 83, 252 CAS.
- Temperatures were calibrated using a standard methanol NMR thermometer (a) A. L. Van Greet, Anal. Chem., 1968, 40, 2227; Search PubMed; (b) A. L. Van Greet, 1970, 42, 679. .
- Y. Cohen and A. Ayalon, Angew. Chem., Int. Ed. Engl., 1995, 34, 816 CrossRef CAS.
- While the dimer of neutral 1 is not observed, its diffusion constant can be estimated by comparison with polyaromatic hydrocarbons of similar size. For this purpose we chose the effective radius, r, to be 3√3V/4π where V is the van der Waals volume.9 By comparison with 9,10-diphenylanthracene (r = 0.412 nm, D = 1.17 × 10−9 m2 s−1) and 1,3,5,7,9-penta-tert-butylcorannulene (r = 0.514 nm, D = 9.89 × 10−10 m2 s−1) and assuming, as in the Stokes–Einstein relation, that D ∝ r−1, the hypothetical dimer of 1 (r = 0.456 nm) would have a diffusion coefficient of 1.09 × 10−9 m2 s−1While the dimer of neutral 1 is not observed, its diffusion constant can be estimated by comparison with polyaromatic hydrocarbons of similar size. For this purpose we chose the effective radius, r, to be 3√3V/4π where V is the van der Waals volume.9 By comparison with 9,10-diphenylanthracene (r = 0.412 nm, D = 1.17 × 10−9 m2 s−1) and 1,3,5,7,9-penta-tert-butylcorannulene (r = 0.514 nm, D = 9.89 × 10−10 m2 s−1) and assuming, as in the Stokes–Einstein relation, that D ∝ r−1, the hypothetical dimer of 1 (r = 0.456 nm) would have a diffusion coefficient of 1.09 × 10−9 m2 s−1.
- L. F. Fieser and M. Fieser , Reagents for Organic Synthesis, Wiley, New York, 1968, p. 1287. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |