Kinetic study of thermolysis of diarylhomonaphthoquinones. Endo/exo substituent and solvent effects
Takumi
Oshima
*,
Kazushi
Tamada
,
Hatsue
Tamura
and
Toshikazu
Nagai
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Machikaneyama 1-16, Toyonaka, Osaka, 560, Japan
First published on 24th December 1999
Abstract
The kinetics of thermal cyclopropane ring-opening of a series of m- and p-substituted endo/exo diphenylbromohomonaphthoquinones 1a–i and the unsubstituted diphenylchlorohomonaphthoquinone 1j have been investigated and compared with biphenyl-2,2′-diylhalogenohomonaphthoquinones 2a,b. The first-order rate constants k/s−1 for 1a–i at 100 °C in toluene increased with the electron-donating ability of the substituents. The kinetic substituent effects were much more pronounced for the exo family than for the endo one and revealed the crucial role of the resonance contribution of diaryl groups; log (k/ko)exo = −1.99σ+ + 0.086 and log (k/ko)endo = −0.784σ+ + 0.002, respectively. The compounds 2a,b thermolyzed very quickly as compared with the corresponding diphenylhalogenohomonaphthoquinones 1e,j. The kinetic solvent effects on the thermolysis of representative compound 1e were so minute that the rates tended to slightly increase with the solvent polarity but decrease with the solvent basicity. These kinetic results were interpreted in terms of a concerted disrotatory ring opening of the incorporated cyclopropane ring.
Introduction
Cyclopropane and its derivatives are fascinating compounds by virtue of their unusual structural, spectroscopic and chemical properties.1 The cyclopropane ring closely resembles the C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C double bond and can interact with neighboring π-electron systems.2 Therefore, the chemical consequence of the cyclopropane ring is highly dependent upon its conformational alignment associated with the conjugation with π- or p-orbitals.3
C double bond and can interact with neighboring π-electron systems.2 Therefore, the chemical consequence of the cyclopropane ring is highly dependent upon its conformational alignment associated with the conjugation with π- or p-orbitals.3
Thermal ring cleavage of cyclopropane rings with labile leaving groups like halogens,4 tosyl group![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 5 and diazonium ion6 has attracted theoretical attention in view of the stereospecific and disrotatory manner of ring-opening, as predicted by the orbital correlation diagram criteria.7
5 and diazonium ion6 has attracted theoretical attention in view of the stereospecific and disrotatory manner of ring-opening, as predicted by the orbital correlation diagram criteria.7
In our series of studies concerning the quinone-fused cyclopropanes, so-called homoquinones,8 we have recently found that thermolysis of diarylbromohomonaphthoquinones 1e and 2a proceeds via ring cleavage of the incorporated cyclopropane ring to provide as primary products, 2-bromo-3-(diphenylmethylene)-2,3-dihydro-1,4-naphthoquinone 3e and 2-(9-bromofluoren-9-yl)-1,4-naphthoquinone 4a, depending on the structural features of the diaryl moieties (Scheme 1).9 These homoquinones are intriguing compounds in that the incorporated cyclopropane rings are highly substituted with π-conjugative endo and exo aromatic nuclei as well as the two quinone carbonyl functions.
 | ||
| Scheme 1 | ||
In this paper, we investigated the substituent and solvent effects on the thermolysis rates of a series of m- and p-substituted endo/exo diphenylhomonaphthoquinones 1a–k as compared with biphenyl-2,2′-diyl-substituted analogues 2a–c in order to gain an insight into the mechanistic pathways for thermolysis of homoquinones.
Results and discussion
Synthesis
Homonaphthoquinones 1a–k were prepared by 1,3-dipolar cycloaddition of m- and p-substituted diphenyldiazomethanes (DDMs) with 2-bromo-1,4-naphthoquinone as described previously.10 The monosubstituted DDMs provided a mixture of endo and exo isomers. Each isomer was separated by column chromatography and purified by recrystallization. The stereochemistry was deduced on the basis of 1H NMR measurements. The endo isomers are characterized by their higher field A2B2 quartet for the p-substituted phenyl ring due to the shielding effects of the facing naphthoquinone aromatic nucleus. For example, the methoxy-substituted endo-1b exhibited the A2B2 quartet at δ 6.44 and 7.06 ppm for the p-anisyl group, while exo-1b did so at δ 6.63 and 7.28 ppm, respectively. The endo/exo differentiating chemical shifts for other substituents are also noticeably large: 0.39–0.46 ppm (1d: p-CH3), 0.38–0.42 (1g: p-Cl) and 0.17–0.41 (1i: p-NO2), respectively (see Experimental section). Biphenyl-2,2′-diylhomonaphthoquinones 2a–c were synthesized by the reaction of 9-diazofluorene (9-DF) with the corresponding naphthoquinones as described elsewhere.11 In the case of the reaction with 2-bromo-1,4-naphthoquinone, 2-(9-bromofluoren-9-yl)-1,4-naphthoquinone (4a) was obtained instead in almost quantitative yield (97%) due to a spontaneous thermolysis of the labile homonaphthoquinone 2a (Scheme 1b).9
Product study
Unsubstituted diphenylbromohomonaphthoquinone 1e thermolyzed at 100 °C in toluene for 24 h to give 2-bromo-3-(diphenylmethylene)-2,3-dihydro-1,4-naphthoquinone 3e in an almost quantitative yield.9 (Scheme 1a). The X-ray crystal structure of 3e is shown in Fig. 1. It is worth noting that the naphthoquinone moiety adopts a non-planar conformation. The plane through the C(3)–C(2)–C(11) linkage significantly flips by 36.6° with respect to the best plane defined by the rest of the naphthoquinone moiety in such a way that the bulky diphenylmethylene function is effectively remote from the Br atom. | ||
| Fig. 1 Molecular structure of 3e. | ||
Further heating of some of the primary pyrolysates 3 resulted in the complication of product analysis owing to the occurrence of several consecutive reactions like radical dimerization, hydrogen abstraction and intramolecular cyclization.9 Therefore, some of the products 3 were confirmed by trapping with methanol to afford SN2′ type adducts 5 (Scheme 2). Unfortunately, thermolysis of CH3O-substituted 1a and 1b yielded complex reaction mixtures (by HPLC and NMR) and afforded no identified products in the methanol trapping experiment. The m-dinitro-substituted 1h provided an intractable yellow powder (see Experimental section). In view of the clean first-order kinetics, these homoquinones 1a, 1b and 1h appear to undergo primary cyclopropane ring cleavage as do others (vide infra).
 | ||
| Scheme 2 | ||
Unlike bromohomonaphthoquinones 1a–i, the chlorohomonaphthoquinone 1j needed a high temperature (150 °C) to practically give the (diphenylmethylene)dihydro-1,4-naphthoquinone 3j which was easily transformed to the methanol adduct 5j (=5e) when treated with methanol. However, methylhomonaphthoquinone 1k resisted thermolysis at 150 °C for over one week. By contrast, biphenyl-2,2′-diylchlorohomonaphthoquinone 2b thermolyzed at relatively low temperature to yield a different type of product, 2-(9-chlorofluorenyl)-1,4-naphthoquinone 4b in quantitative yield,9 although the analogous methylhomonaphthoquinone 2c remained intact even after being heated for 1 week at 150 °C (Scheme 1b).9 For 2b, the change of the reaction pathway may be due to the severe steric repulsion between the fluorene peri-hydrogen and the facing carbonyl group for the expected 2-halogeno-3-fluorenylidene-2,3-dihydro-1,4-naphthoquinones 6.9
It is noted here that thermolysis of both the chloro-substituted endo-1g and exo-1g in toluene at 100 °C provided the identical isomer ratio (1∶1.3) of E- and Z-3g (by NMR in CDCl3 at 20 °C). Such a stereo-randomization apparently contradicts the well-known criterion of stereospecificity in the thermal ring-opening of halogenocyclopropanes.7 However, this conflicting phenomenon can be rationalized by the observation that the isolated major isomer of 3g easily isomerized to the minor one even at 50 °C in [2H6]benzene and came to equilibrium at the same isomer ratio of 1∶1.3 as the above thermolysis. This ratio corresponds to the free energy difference (ΔG) of only 0.62 kJ mol−1 at 50 °C. The PM3 calculation predicted that the E structure is somewhat more stable than the Z one by only 0.6 kJ mol−1 in harmony with the experimental isomer ratio.12 Thus, we tentatively assign the stable isomer as adopting the E form (see Experimental section). The isomerization of the E isomer was moderate in nonpolar [2H6]benzene (k = 2.17 × 10−4 s−1 and t1/2 = 0.89 h at 50 °C), but too fast to be followed by NMR in polar [2H3]acetonitrile. Hence, the thermal E–Z isomerization seems to occur via a resonance stabilized zwitterionic intermediate I.

Kinetic study
a
| Entry | Homoquinone | X1 | X2 | X3 | 106k/s−1b |
|---|---|---|---|---|---|
| a Carried out in sealed capillary tubes. b The k values are the average of at least two measurements. Error limit of k is ±2%. c Extrapolated value from the k values at higher temperature, 5.58(±0.084) × 10−5 s−1 (150 °C) and 1.99(±0.035) × 10−4 s−1 (170 °C), respectively. d No reaction over 1 week heating at 100 °C. e Spontaneously decomposed in situ on preparation of homoquinone 2a at room temperature. | |||||
| 1 | 1a | p-OCH3 | p-OCH3 | Br | 6260 ± 94 |
| 2 | endo-1b | p-OCH3 | H | Br | 171 ± 3.0 |
| 3 | exo-1b | H | p-OCH3 | Br | 2360 ± 33 |
| 4 | 1c | p-CH3 | p-CH3 | Br | 359 ± 4.7 |
| 5 | endo-1d | p-CH3 | H | Br | 87.2 ± 1.1 |
| 6 | exo-1d | H | p-CH3 | Br | 199 ± 2.6 |
| 7 | 1e | H | H | Br | 46.2 ± 0.51 |
| 8 | 1f | p-Cl | p-Cl | Br | 27.6 ± 0.39 |
| 9 | endo-1g | p-Cl | H | Br | 40.3 ± 0.60 |
| 10 | exo-1g | H | p-Cl | Br | 37.2 ± 0.57 |
| 11 | 1h | m-NO2 | m-NO2 | Br | 0.805 ± 0.010 |
| 12 | endo-1i | p-NO2 | H | Br | 10.3 ± 0.13 |
| 13 | exo-1i | H | p-NO2 | Br | 1.64 ± 0.025 |
| 14 | 1j | H | H | Cl | 0.946 ± 0.014c |
| 15 | 1k | H | H | CH3 | NRd |
| 16 | 2a | H | H | Br | very faste |
| 17 | 2b | H | H | Cl | 1390 ± 19 |
| 18 | 2c | H | H | CH3 | NRd |
The linear free energy treatment of log (k/ko) for the respective mono-substituted endo and exo isomers gave a better fit versus Brown σ+13 than Hammett σ,14 where ko is the rate constant for the unsubstituted 1e (Fig. 2). The regression equations are log (k/k0)exo = −1.99σ+ + 0.086 (r = 0.998, s = 0.090, n = 5) for the exo series and log (k/k0)endo = −0.784σ+ − 0.002 (r = 0.997, s = 0.043, n = 5) for the endo one, respectively. For σ parameters, rather worse correlations are obtained for both series due to the noticeable upper deviation of the CH3O substituent; log (k/ko)exo = −2.60σ + 0.455 (r = 0.942, s = 0.45, n = 5) and log (k/ko)endo = −1.06σ + 0.147 (r = 0.972, s = 0.12, n = 5), respectively. The excellent correlation against σ+ with negative ρ values suggests that electron-donating resonance effects of the substituent play an important role in stabilizing the transition state, although a limited set of present data is not adequate for complete knowledge of the substituent effects. The absolute ρ value for exo isomers is about 2.5 times as large as that for endo isomers. This means a preferential effect of the exo aromatic substituents on the cyclopropane ring-cleavage of homonaphthoquinones. Also of interest is that the endo/exo disubstituted homonaphthoquinones with identical substituents (X1 = X2) gave an excellent correlation when plotted against the sum of the σ+ values: log (k/k0)disub = −1.34σ+ + 0.046 (r = 1.00, s = 0.029, n = 5). The absolute ρ value is somewhat smaller than half of the sum (=−2.77) of the individual absolute ρ values for the exo and endo monosubstituted series. It is well known that the substituent effects of two aromatic rings are not additive and the discrepancy is most serious when the first substituent is strongly electron-donating or -withdrawing as in the present system.15
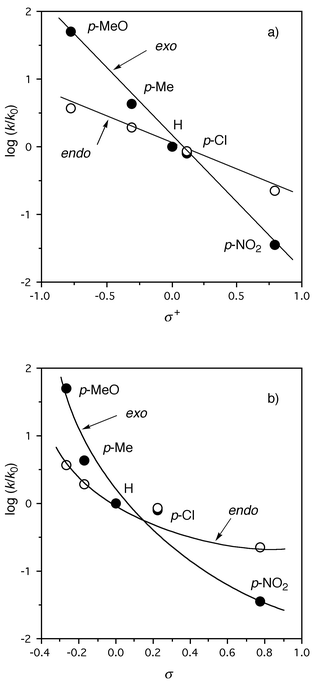 | ||
| Fig. 2 The plots of log (k/k0) for the thermolysis of diarylhomonaphthoquinones in toluene at 100 °C against the a) σ+ and b) σ values; the lines were drawn for the exo-substituted (•) and the endo-substituted (○) series. | ||
In order to obtain some steric features of these aromatic nuclei, we resorted to the X-ray crystal structure of the representative compound 1e.16 The obtained structure shows that there is a significant difference in the rotational freedom between the two phenyl rings. The endo phenyl ring lies in an almost castanets-like conformation with respect to the naphthoquinone plane to minimize the steric repulsion, therefore its rotation appears to be highly restricted. In contrast, the exo phenyl ring is located in the less hindered space. As such, the exo aromatic ring would enjoy a more favorable bisected conformation (II) which is essential for the ideal π-conjugation between the cyclopropane ring and the adjacent π-systems.2,17
As to the effect of the cyclopropane substituent, the less labile chlorine substituent markedly decreased the rate to about 1/50 times slower than the bromine substituent (entries 7 and 14). Of interest is that the replacement of the diphenyl group by planar biphenyl-2,2′-diyl brought about ca. 1500 times rate enhancement (entries 14 and 17). This is probably due to the inherent bisected structure (III) associated with the spiro-linkage of the planar fluorenylidene function to the rigid homoquinone skeleton for 2b.
 18 or solvent basicity (Dπ)19 was insufficient: log k = 0.0195ET − 4.96 (r = 0.759, s = 0.11, n = 10, except acetic acid) and log k = −0.249Dπ − 4.20 (r = 0.863, s = 0.099, n = 8), respectively. To gain more insight into the solvent effects, the two parameter procedure was used to greatly improve the correlation for the solvents for which both parameters are known: log k = 0.0196ET − 0.146Dπ − 4.97 (r = 0.947, s = 0.077, n = 8). The weightings of the ET and Dπ parameters on the regression equation are approximately equal as estimated from their contributions, 54 and 46%, respectively. The small positive coefficient of ET may be due to the rate-acceleration by solvation of a slightly polar transition state in which the fission of the polar C–Br bond will be facilitated in the polar medium. In contrast, the small negative coefficient of Dπ suggests rate-retardation by solvation of the ground state naphthoquinone moiety, since the Dπ parameter can be successfully used to reflect the π-acceptor properties of substrates.20 Consequently, negligible solvent effects, consistent with the very poor polarization in the transition state, support a concertedness of the present thermolysis, indicating that the ring opening is synchronous with the departure of the leaving bromide.
a
18 or solvent basicity (Dπ)19 was insufficient: log k = 0.0195ET − 4.96 (r = 0.759, s = 0.11, n = 10, except acetic acid) and log k = −0.249Dπ − 4.20 (r = 0.863, s = 0.099, n = 8), respectively. To gain more insight into the solvent effects, the two parameter procedure was used to greatly improve the correlation for the solvents for which both parameters are known: log k = 0.0196ET − 0.146Dπ − 4.97 (r = 0.947, s = 0.077, n = 8). The weightings of the ET and Dπ parameters on the regression equation are approximately equal as estimated from their contributions, 54 and 46%, respectively. The small positive coefficient of ET may be due to the rate-acceleration by solvation of a slightly polar transition state in which the fission of the polar C–Br bond will be facilitated in the polar medium. In contrast, the small negative coefficient of Dπ suggests rate-retardation by solvation of the ground state naphthoquinone moiety, since the Dπ parameter can be successfully used to reflect the π-acceptor properties of substrates.20 Consequently, negligible solvent effects, consistent with the very poor polarization in the transition state, support a concertedness of the present thermolysis, indicating that the ring opening is synchronous with the departure of the leaving bromide.
a
| Solvent | 105k/s−1b |
E
Tc |
D
πd |
|---|---|---|---|
|
a
Carried out in sealed capillary tubes.
b
The k values are the average of at least two measurements. Error limit of k is ±2%.
c
ET values; see ref. 18.
d
Dπ values; see ref. 19.
e
The k values at 110 and 120 °C are 2.54(±0.03) × and 6.74(±0.10) × 10−4 s−1, respectively. The activation parameters are ΔH‡ = 120.3(±0.2) kJ mol−1 and ΔS‡ = −2.5(±0.5) J mol−1 K−1 at 100 °C, respectively.
|
|||
| Nitromethane | 12.3 ± 0.18 | 46.3 | −0.724 |
| 1,2-Dichloroethane | 10.2 ± 0.14 | 41.3 | −1.22 |
| Acetonitrilee |
8.43 ± 0.16 | 45.6 | −0.440 |
| Butan-2-one | 8.37 ± 0.11 | 41.3 | 0.177 |
| Ethanol | 8.30 ± 0.13 | 51.9 | — |
| Benzene | 5.27 ± 0.05 | 34.3 | 0 |
| Triethylamine | 5.26 ± 0.07 | 32.1 | — |
| Toluene | 4.62 ± 0.08 | 33.9 | 0.394 |
| Ethyl acetate | 4.56 ± 0.06 | 38.1 | 0.289 |
| 1,4-Dioxane | 4.31 ± 0.08 | 36.0 | 0.590 |
| Acetic acid | 2.61 ± 0.05 | 51.7 | — |
Mechanistic considerations
It is generally accepted that thermolysis of cyclopropanes with labile leaving groups proceeds through a disrotatory mode of ring-opening to provide the propene derivatives.7 This cyclopropyl–allyl cationic rearrangement is concerted and the stereochemistry can thus be predicted on the basis of the principle of orbital symmetry conservation.7According to this mechanistic pathway, the present kinetic substituent effects, which exhibited a significant contribution of the exo substituents (Table 1), can be rationalized by considering that the electron-donating exo aryl group rotates inward in such a way that the lobes of the opening adjacent C2–C3 bond will facilitate the departure of the leaving bromide from the C1 atom and stabilize the developing positive charge. The fact that the logarithmic rate constants correlate with σ+, rather than σ, provides strong evidence for the resonance stabilization of the positive charge by the exo aromatic nucleus. Such a stereoelectronic interaction is at a maximum when the p-orbital axis of the aromatic ring is arranged parallel to the cyclopropane ring (bisected conformation). In fact, it has become apparent from the X-ray crystal structure of 1e16 that the less restricted exo aromatic ring is more easily capable of adopting a favorable bisected conformation so as to resonate with the breaking cyclopropane hybrid bond.17 Such a preferable π-conjugation is less likely for the endo aromatic ring because of the highly congested circumstances. Moreover, as shown in Table 2, the poor kinetic solvent effects strongly support the concerted cyclopropane bond-opening linked with a simultaneous bromide migration. The small negative entropy of activation for the parent 1e signifies a reactant-like transition state (ΔS‡ = −2.5 (0.5) J mol−1 K−1 and ΔH‡ = 120.3(0.2) kJ mol−1 at 100 °C in acetonitrile, cf.Table 2).
The present kinetic results can also be interpreted by analyzing interactions of frontier orbitals. In this approach, the orbital interaction [σ2s + ω0s] can be envisaged,21 in which the σ orbital (HO) of the C2–C3 bond interacts with the central vacant p orbital (LU), designated as ω. The electron donation from the aryl group will raise the HO energy and thereby enhance the overlapping with the LU orbital. An alternative orbital interaction [σ2s + σ2a] can also satisfactorily explain our experimental results without postulating an intervention of cyclopropyl cation.22 As illustrated in Fig. 3, the thermally allowed concerted process involves charge transmittance from the cyclopropane C2–C3 bond (HO) to the antibonding C1–Br bond (LU). In the disrotatory motion, the electron donation from the bisected exo-aryl group will mostly raise the HO energy and facilitate the bromide migration to the C3 atom (path a) for diphenyl-substituted series or the C2 atom (path b) for biphenyl-2,2′-diyl-substituted ones, respectively. In contrast, the endo aryl group cannot satisfactorily participate in the orbital interaction because of the insufficient bisected conformation. The rapid thermolysis for biphenyl-2,2′-diylhomonaphthoquinones is ascribed to the intrinsic bisected structure in which the planar fluorenylidene function can enjoy the ideal resonance interaction with the quasi π-orbital of cyclopropane ring. The path b migration is thought to be the result of a serious steric repulsion between the peri-hydrogen of the fluorenylidene function and the adjacent carbonyl group.9 Whatever the mechanism, the present thermolysis of homoquinones proceeded through a concerted disrotatory ring-opening, undergoing the marked resonance effects of the exo aryl group.
![[σ2s + σ2a] orbital interaction.](/image/article/2000/P2/a905223b/a905223b-f3.gif) | ||
| Fig. 3 [σ2s + σ2a] orbital interaction. | ||
Conclusion
It was found that thermolysis of a series of m- and p-substituted endo/exo diphenylbromohomonaphthoquinones 1a–i, which undergo cyclopropane ring-cleavage to provide the corresponding 2-bromo-3-diphenyl-2,3-dihydro-1,4-naphthoquinones as primary pyrolysates, was much more accelerated by electron-donating substituents in the exo aromatic rings than in the endo ones, as indicated by the Hammett ρ values of −1.99 and −0.784 against σ+, respectively. However, the solvent effect on thermolysis of the representative compound 1e was so small that the rate constant increased by only a factor of several times in spite of the wide range of solvent polarity. It was also noticed that due to the intrinsic bisected conformation biphenyl-2,2′-diylhalogenohomonaphthoquinones 2a,b thermolyzed more quickly to afford 2-(9-halogenofluoren-9-yl)-1,4-naphthoquinones 4a,b instead of the predicted dihydro-1,4-naphthoquinones. These results were rationalized on the basis of a concerted disrotatory ring opening associated with the resonance interaction of the endo and exo aryl groups.Experimental
Melting points were measured on a Yanagimoto microscopic apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a JEOL EX-270 MHz spectrometer with Me4Si as an internal standard and CDCl3 as solvent unless otherwise noted. IR spectra were taken with a Perkin-Elmer 938G spectrophotometer. Mass spectra were obtained on a JEOL JMS DX303 spectrometer.Kinetic measurements
The kinetic measurements were carried out on a Hitachi 655A-12 liquid chromatograph equipped with a Waters RCM 8 × 10 module, installing a Radial-Pak cartridge (8C18, 5μ) in a similar manner to that previously described.9 The reaction solutions for thermolysis of variously substituted 1a–k and 2b,c were prepared by mixing the homoquinone and naphthalene or biphenyl (as an internal standard) in toluene at ordinary temperature and sealed in 10–15 glass capillaries (ϕ = 1.8 mm). The reaction solutions for the kinetic solvent effects were similarly prepared by mixing unsubstituted 1e and the internal standard in a given solvent and sealed in the capillary tubes. The reactions were initiated by fast immersion into a Haake EF thermostatted bath (±0.05 °C) at the given temperatures and after several minutes the reaction was followed at time intervals by monitoring the decreasing relative integral absorptions of homoquinone versus the internal standard at 280 nm up to the conversion of second-half lives. Clear first-order kinetics were observed for all reactions studied by plotting logarithmic values of the relative absorptions against time.Materials
All solvents used were dried and purified in the usual manner.23 All homoquinones 1a–k and 2b,c were prepared from the reactions of variously substituted diphenyldiazomethanes (DDMs) and 9-diazofluorene (9-DF) with 2-bromo-, 2-chloro-, and 2-methyl-1,4-naphthoquinones according to the previous procedure.10 These homonaphthoquinones were isolated by column chromatography on silica gel and purified by recrystallization from a mixture of hexane and benzene. All new compounds 1a–d and 1f–i provided satisfactory analytical and spectroscopic data as shown below. The known compounds 1e,91j,241k25 and 2b,242c25 were reported elsewhere. The compound 2a was not obtained because of its thermal lability leading to ring-cleaved 4a.9
![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) p-anisyl)-3,4-benzobicyclo[4.1.0]heptane-2,5-dione 1a..
Yield 55% (isolated); mp 126–128 °C; νmax/cm−1 1685, 1606, 1510, 1289, 1250, 1179, 1029 and 831; δH 3.55 (3H, s), 3.78 (3H, s), 3.88 (1H, s), 6.44 (2H, d, J 8.91 Hz), 6.87 (2H, d, J 8.58 Hz), 7.03 (2H, d, J 8.58 Hz), 7.40 (2H, d, J 8.91 Hz), 7.45–7.53 (2H, m), 7.80–7.87 (1H, m) and 7.87–7.94 (1H, m); m/z (EI) 383 (M+ − Br). (Found: C, 65.02; H, 4.31. C25H19O4Br requires C, 64.80; H, 4.13%).
p-tolyl)-3,4-benzobicyclo[4.1.0]heptane-2,5-dione 1c..
Yield 68% (isolated); mp 134–135 °C; νmax/cm−1 1678, 1591, 1510, 1286, 812, 774, 718 and 685; δH 2.00 (3H, s), 2.30 (3H, s), 3.76 (1H, s), 6.70 (2H, d, J 7.92 Hz), 7.01 (2H, d, J 7.92 Hz), 7.15 (2H, d, J 7.92 Hz), 7.39 (2H, d, J 7.92 Hz), 7.43–7.51 (2H, m), 7.77–7.85 (1H, m) and 7.85–7.93 (1H, m); m/z (EI) 430 (M+). (Found: C, 69.73; H, 4.60. C25H19O2Br requires C, 69.61; H, 4.44%).
p-anisyl)-3,4-benzobicyclo[4.1.0]heptane-2,5-dione 1a..
Yield 55% (isolated); mp 126–128 °C; νmax/cm−1 1685, 1606, 1510, 1289, 1250, 1179, 1029 and 831; δH 3.55 (3H, s), 3.78 (3H, s), 3.88 (1H, s), 6.44 (2H, d, J 8.91 Hz), 6.87 (2H, d, J 8.58 Hz), 7.03 (2H, d, J 8.58 Hz), 7.40 (2H, d, J 8.91 Hz), 7.45–7.53 (2H, m), 7.80–7.87 (1H, m) and 7.87–7.94 (1H, m); m/z (EI) 383 (M+ − Br). (Found: C, 65.02; H, 4.31. C25H19O4Br requires C, 64.80; H, 4.13%).
p-tolyl)-3,4-benzobicyclo[4.1.0]heptane-2,5-dione 1c..
Yield 68% (isolated); mp 134–135 °C; νmax/cm−1 1678, 1591, 1510, 1286, 812, 774, 718 and 685; δH 2.00 (3H, s), 2.30 (3H, s), 3.76 (1H, s), 6.70 (2H, d, J 7.92 Hz), 7.01 (2H, d, J 7.92 Hz), 7.15 (2H, d, J 7.92 Hz), 7.39 (2H, d, J 7.92 Hz), 7.43–7.51 (2H, m), 7.77–7.85 (1H, m) and 7.85–7.93 (1H, m); m/z (EI) 430 (M+). (Found: C, 69.73; H, 4.60. C25H19O2Br requires C, 69.61; H, 4.44%).
Thermolysis of 1a–j
Preparative thermolysis was carried out at 100 °C in a sealed benzene solution according to the method employed previously for 1e.9 We succeeded in isolating the primary ring-opened products 3c, 3f, and 3g (major isomer) by recrystallization from the reaction mixture. However, the thermolysis of 1a, endo-, exo-1b and 1h provided a complicated product mixture probably due to several subsequent reactions. So in confirming that the thermolysis primarily produces the allylic halides 3, we carried out a similar thermolysis in the presence of the additive methanol (10% by volume) and obtained the corresponding methanol adducts 5c,d and 5f,g, and 5i (Scheme 2). Unfortunately, this trapping experiment still failed to give the corresponding methanol adducts for the methoxy-substituted 1a,1b, and the m-dinitro-substituted 1h. The compounds 1a and 1b yielded dark intractable resinous products, while 1h afforded an insoluble and inseparable yellow powder. Thermolysis of 1j (50 mg) was performed at 150 °C for 7 h in a sealed toluene solution (0.5 ml) to yield an almost quantitative amount of 3j (by NMR). When treated with methanol, 3j was easily transformed to the methanol adduct 5j (= 5e). The spectral data of the isolated pyrolysates and the methanol adducts are as follows. The analytical data for 5e and 4b are given elsewhere.9Crystal structure determination of compound 3e†
2) = 0.170 for all 5133 unique reflections. Intensity data in the θ range 3–30° were corrected on a Rigaku AFC5R diffractometer with graphite-monochromated Mo-Kα radiation and corrected for Lorentz and polarization effects, and for absorption. The structure was solved by direct methods and refined on F2 by full-matrix least-squares methods anisotropically for non-hydrogen atoms. All hydrogen atoms fixed at magnitudes in excess of 1 e Å−3 were observed on the difference-Fourier maps.
References
- (a) The Chemistry of the Cyclopropyl Group, part 1 and 2, ed. Z. Rappoport, Wiley, New York, 1987; Search PubMed; (b) The Chemistry of the Cyclopropyl Group Volume 2, ed. Z. Rappoport, Wiley, New York, 1995; Search PubMed; (c) H. N. C. Wong, M. Y. Hon, C. W. Tse and Y. C. Yip, Chem. Rev., 1989, 89, 165 CrossRef CAS.
- (a) M. Charton , The Chemistry of the Alkenes, 2, ed. J. Zabicky, Interscience, London, ch. 10, 1970; Search PubMed; (b) A. de Meijere, Angew. Chem., Int. Ed. Engl., 1979, 18, 809 CrossRef.
- (a) T. T. Tidwell , The Chemistry of the Cyclopropyl Group, part 2, ed. Z. Rappoport, ch. 10, pp. 565–632, Wiley, New York, 1987; Search PubMed; (b) T. A. Crabb and A. V. Patel , Rodd’s Chemistry of Carbon Compounds, 2nd Suppl. to the 2nd edn., ed. M. Sainsbury, Elsevier, Amsterdam, vol. IIA/B, ch. 1, pp. 1–80, 1992. Search PubMed.
-
(a) D. C. Duffey, J. P. Minyard and R. H. Lane, J. Org. Chem., 1966, 31, 3865 CAS;
(b) L. Ghosez, P. Laroche and G. Slinckx, Tetrahedron Lett., 1967, 2767 CrossRef CAS;
(c) M. S. Baird and C. B. Reese, Tetrahedron Lett., 1967, 1379 CrossRef CAS;
(d) C. W. Jefford, E. H. Yen and R. Medary, Tetrahedron Lett., 1966, 2069 CrossRef CAS;
(e) P. v. R. Schleyer, T. M. Su, M. Saunder and J. C. Rosenfeld, J. Am. Chem. Soc., 1969, 91, 5174 CrossRef CAS;
(f) M. S. Baird, D. G. Lindsay and C. B. Rees, J. Chem. Soc. (C), 1969, 1173 Search PubMed;
(g) D. C. Horwell and C. W. Rees, J. Chem. Soc., Chem. Commun., 1969, 1428 Search PubMed;
(h) C. B. Reese and A. Shaw, J. Am. Chem. Soc., 1970, 92, 2566 CrossRef CAS;
(i) M. S. Baird, B. S. Mahli and L. Sheppard, J. Chem. Soc., Perkin Trans. 1, 1990, 1881 RSC.
- (a) C. H. Depuy, L. G. Schnack, J. W. Hausser and W. Wiedemann, J. Am. Chem. Soc., 1965, 87, 4006 CrossRef CAS; (b) C. H. Depuy, L. G. Schnack and J. W. Hausser, J. Am. Chem. Soc., 1966, 88, 3343 CrossRef CAS; (c) P. v. R. Schleyer, G. W. Van Dine, U. Schöllkopfand and J. Paust, J. Am. Chem. Soc., 1966, 88, 2868 CrossRef CAS.
- W. Kirmse and H. Schütte, J. Am. Chem. Soc., 1967, 89, 1284 CrossRef CAS.
- (a) R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395 CrossRef CAS; (b) C. H. Depuy, Acc. Chem. Res., 1968, 1, 33 CrossRef CAS; (c) L. Radom, P. C. Hariharan, J. A. Pople and P. v. R. Schleyer, J. Am. Chem. Soc., 1973, 95, 6531 CrossRef CAS.
- (a) T. Oshima and T. Nagai, Tetrahedron Lett., 1993, 34, 649 CrossRef CAS; (b) T. Oshima, Y. Nakajima and T. Nagai, Chem. Lett., 1993, 1977 CAS; (c) H. Moriwaki, T. Oshima and T. Nagai, J. Chem. Soc., Chem. Commun., 1994, 255 RSC; (d) H. Moriwaki, T. Oshima and T. Nagai, J. Chem. Soc., Chem. Commun., 1994, 1681 RSC; (e) H. Moriwaki, T. Oshima and T. Nagai, J. Chem. Soc., Perkin Trans. 1, 1995, 2517 RSC; (f) H. Moriwaki, K. Fukushima, T. Nagai and T. Oshima, Chem. Commun., 1996, 495 RSC; (g) H. Moriwaki, T. Matsumoto, T. Nagai and T. Oshima, J. Chem. Soc., Perkin Trans. 1, 1996, 1461 RSC.
- T. Oshima, K. Tamada and T. Nagai, J. Chem. Soc., Perkin Trans. 1, 1994, 3325 RSC.
- T. Oshima and T. Nagai, Bull. Chem. Soc. Jpn., 1988, 61, 2507 CAS.
- T. Oshima and T. Nagai, Bull. Chem. Soc. Jpn., 1989, 62, 2580 CAS.
- The calculations by the PM3 method were performed with the MOPAC program (ver. 6) using a Cache Work-System.
- (a) H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1957, 79, 1913 CrossRef CAS; (b) ibid., 1958, 80, 4979; Search PubMed; (c) ibid., 1959, 81, 3323. Search PubMed.
- (a) H. H. Jaffé, Chem. Rev., 1953, 53, 191 CrossRef CAS; (b) D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420 CrossRef CAS.
- Y. Tsuno and M. Fujino, Adv. Phys. Org. Chem., 1999, 32, 267 Search PubMed.
- T. Oshima , K. Fukushima and T. Kawamoto , Acta Crystallogr., Sect. C, 1999, 55, 608. Search PubMed.
- The C–C bond hybridization of the cyclopropane ring is popularly envisioned by the Walsh model (cf. A. D. Walsh, Nature, 1947, 165, 712 Search PubMed; A. D. Walsh, Trans. Faraday Soc., 1949, 45, 179 ); the lowest energy orbital (σ) by a linear combination of the three sp2-hybrid atomic orbitals (AOs) and the two equal-energy orbitals (eS and eA, so-called quasi π) by linear combinations of three p-orbitals. Accordingly, the conjugation energy is a maximum when the neighboring p-orbital axis is arranged parallel to the plane of the cyclopropane ring (bisected conformation), while the magnitude of interaction is a minimum for the perpendicular arrangement.
- (a) C. Reichardt, Angew. Chem., Int. Ed. Engl., 1979, 18, 98 CrossRef; (b) C. Reichart and E. H. Görnert, Liebigs Ann. Chem., 1983, 721 Search PubMed; (c) C. Reichart, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- T. Oshima , S. Arikata and T. Nagai , J. Chem. Res., 1981, (S) 204, (M) 2518. Search PubMed.
- (a) P. Brown and R. C. Cookson, Tetrahedron, 1965, 21, 1977 CrossRef CAS; (b) T. Oshima and T. Nagai, Bull. Chem. Soc. Jpn., 1982, 55, 555 CAS; (c) T. Oshima and T. Nagai, Tetrahedron Lett., 1985, 26, 4785 CrossRef CAS; (d) T. Oshima and T. Nagai, J. Chem. Soc., Chem. Commun., 1994, 2787 RSC.
- E. L. Ansari , R. Qureshi and M. L. Qureshi , Electrocyclic Reactions, Wiley-VCH, Weinheim, 1999. Search PubMed.
- K. Fukui, Acc. Chem. Res., 1971, 4, 57 CrossRef CAS.
- J. A. Riddick and W. B. Bunger , Techniques of Chemistry, Vol. II, Organic Solvents, 3rd edn., ed A. Weissberger, John Wiley, New York, 1970. Search PubMed.
- Y. Nakano, M. Hamaguchi and T. Nagai, J. Org. Chem., 1989, 54, 1135 CrossRef CAS.
- F. M. Dean, P. G. Jones, R. B. Morton and P. S. Sidisunthorn, J. Chem. Soc., 1963, 5336 RSC.
Footnote |
| † CCDC reference number 188/191. See http://www.rsc.org/suppdata/p2/a9/a905223b for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2000 |