Crystal structures and solid-state reactivities of 1,4- and 1,2-bis(5-hydroxypenta-1,3-diynyl)benzenes and 1-(5-hydroxypenta-1,3-diynyl)-4-ethynylbenzene
Eiko
Mochizuki
*a,
Yoshiyuki
Shibamoto
a,
Kazuma
Yano
a,
Nobuko
Kanehisa
a,
Yasushi
Kai
a,
Seiichi
Tagawa
b and
Yukio
Yamamoto
b
aDepartment of Materials Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka, 565-0871, Japan
bThe Institute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka, 567-0047, Japan
First published on 14th January 2000
Abstract
Crystal structures and the solid-state polymerization of 1,4-bis(5-hydroxypenta-1,3-diynyl)benzene (1), 1,2-bis(5-hydroxypenta-1,3-diynyl)benzene (2) and 1-(5-hydroxypenta-1,3-diynyl)-4-ethynylbenzene (3) were investigated. The crystal structure of 1 depends on the recrystallization solvent. Crystals obtained from acetone–benzene mixtures polymerize when irradiated with γ-rays or heated. The colourless crystals become black and are insoluble in common organic solvents. Only one of the butadiynyl groups participates in the radiation-induced polymerization, whereas both do, in the thermal polymerization. Polymerization does not occur for crystals obtained from pure acetone. The polymerization reactivities are correlated to the crystal structures. Polymerizable crystals were not obtained from 2. The arrangement of the butadiynyl groups in crystals of 2 is unfavorable for the polymerization. Crystals of 3, obtained from hexane–acetone mixtures, polymerize upon γ-irradiation but not upon heating. In the radiation-induced polymerization of 3, the propagation by the ethynyl and butadiynyl groups is considered to proceed independently along different crystal axes. The ethynyl groups are more reactive than the butadiynyl groups because they have a shorter intermolecular carbon–carbon distance through which the polymerization occurs. The difference in reactivity toward heating between 1 and 3 may also be interpreted in terms of intermolecular carbon–carbon distances of the butadiynyl groups.
Introduction
We have previously reported on the reactions of γ-irradiated crystals of diethynylbenzene derivatives.1–3 The crystal structures of the monomers were markedly affected by the nature of the substituents and their positions in the aromatic ring, leading to a variety of solid-state reactivities. Polymerization or dimerization occurred selectively depending on the crystal structures in contrast to the non-selective reactivities in solution. The present study is concerned with the crystal structures and solid-state polymerization of bifunctional derivatives having diacetylene moieties. The structures and the melting points of the monomers, 1,4-bis(5-hydroxypenta-1,3-diynyl)benzene (1), 1,2-bis(5-hydroxypenta-1,3-diynyl)benzene (2) and 1-(5-hydroxypenta-1,3-diynyl)-4-ethynylbenzene (3), used in the present study, are shown in Fig. 1. | ||
| Fig. 1 Structures of monomers. | ||
Solid-state polymerization of diacetylene derivatives has been extensively studied in connection with both the propagation processes and the π-conjugation properties of the product polymers, including nonlinear optical properties.4–6 Recently, attempting the enhancement of π-conjugation properties, bifunctional diacetylene monomers and octatetraynes have been synthesized and their polymerization reactivities have been investigated.7–14 Radiation-induced solid-state polymerization has been reported for 1,4-bis(octadeca-1,3-diynyl)benzene, an analogue of 1. However, there has been no report on the crystal structures of bifunctional diacetylene monomers. In the present study the crystallographic analysis was undertaken to elucidate the contribution of the two functional groups to the polymerization.
The crystal structure of 1 depended on recrystallization solvents. Changes in the recrystallization conditions of 1 revealed that it exists in two crystalline forms, designated as 1a and 1b. 1a Crystals were obtained from acetone–benzene mixtures and 1b crystals, from pure acetone. Upon γ-irradiation, 1a polymerized so as to be insoluble in common organic solvents. Polymers were obtained neither from 1b, nor from the crystals of 2. Reactivities in polymerization are correlated to the monomer packings in the crystals. Polymerization was also examined for 3.
Experimental
Polymerization experiments
The monomers, 1 and 3, were prepared by the coupling of 1,4-diethynylbenzene and 3-bromopropyn-1-ol by the literature method![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 15 and were purified by repeated recrystallization. Similarly, 2 was prepared from 1,2-diethynylbenzene and 3-bromopropyn-1-ol and was recrystallized. The diethynylbenzenes were prepared from 1,2- and 1,4-diiodobenzenes (Aldrich and Wako Chemicals, respectively) according to the literature.16 3-Bromopropyn-1-ol was prepared according to the literature.17 The monomer crystals were sealed in Pyrex tubes under vacuum and were irradiated at room temperature with γ-rays from a 60Co source at a dose rate of 5.2 kGy h−1 (1 Gy = 1 J kg−1). The solid-state polymerization was followed by comparing the relative intensities of the stretching bands of the corresponding carbon–carbon triple bonds, –C
15 and were purified by repeated recrystallization. Similarly, 2 was prepared from 1,2-diethynylbenzene and 3-bromopropyn-1-ol and was recrystallized. The diethynylbenzenes were prepared from 1,2- and 1,4-diiodobenzenes (Aldrich and Wako Chemicals, respectively) according to the literature.16 3-Bromopropyn-1-ol was prepared according to the literature.17 The monomer crystals were sealed in Pyrex tubes under vacuum and were irradiated at room temperature with γ-rays from a 60Co source at a dose rate of 5.2 kGy h−1 (1 Gy = 1 J kg−1). The solid-state polymerization was followed by comparing the relative intensities of the stretching bands of the corresponding carbon–carbon triple bonds, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H (3263 cm−1) and –CC–CC– (2240 cm−1), with those of the aromatic rings (832 cm−1) of the FTIR spectra (KBr) of the original and irradiated crystals. The FTIR spectra were recorded on a Perkin-Elmer Spectrum 2000 spectrophotometer. Fig. 2 shows the FTIR spectra of the original crystals of 1a and 3, which polymerized upon γ-irradiation or heating. Gel permeation chromatographic measurement was carried out by 262 nm absorption in tetrahydrofuran using a Shimadzu CR3A equipped with two 30 cm TSK-Gel G6000PWXL and G3000PWXL columns connected in series.
C–H (3263 cm−1) and –CC–CC– (2240 cm−1), with those of the aromatic rings (832 cm−1) of the FTIR spectra (KBr) of the original and irradiated crystals. The FTIR spectra were recorded on a Perkin-Elmer Spectrum 2000 spectrophotometer. Fig. 2 shows the FTIR spectra of the original crystals of 1a and 3, which polymerized upon γ-irradiation or heating. Gel permeation chromatographic measurement was carried out by 262 nm absorption in tetrahydrofuran using a Shimadzu CR3A equipped with two 30 cm TSK-Gel G6000PWXL and G3000PWXL columns connected in series.
 | ||
| Fig. 2 FTIR spectra of the original crystals of (A) 1a and (B) 3. | ||
X-Ray crystal structure analysis
The crystal data and experimental details are summarized in Table 1. X-Ray diffraction data were collected by a Rigaku AFC-5R diffractometer with graphite-monochromated Cu-Kα radiation (λ = 1.54178 Å) up to 2θmax of 120.1°. All the crystallographic calculations were carried out by using the TEXSAN software package of the Molecular Structure Corporation. The crystal structures were solved by the direct method (SHELXS 86) and were refined by full-matrix least-squares. All the non-hydrogen atoms and hydrogen atoms were refined anisotropically and isotropically, respectively.| Parameter | 1a | 1b | 2 | 3 |
|---|---|---|---|---|
| a R = Σ(|Fo| − |Fc|)/Σ|Fo|. b Rw = [Σω(|Fo| − |Fc|)2/Σω|Fo|2]1/2. c GOF = [Σω(|Fo| − |Fc|)2/(m − n)]1/2. Diffractometer: Rigaku AFC-5R. Crystal structure analysis: TEXSAN. Structure solution: direct method (SHELXS86). Refinement: full-matrix least-squares. | ||||
| Empirical formula | C16H10O2 | C16H10O2 | C16H10O2 | C13H8O |
| Formula weight | 234.25 | 234.25 | 234.25 | 180.21 |
| Crystal dimensions/mm | 0.30 × 0.25 × 0.10 | 0.30 × 0.20 × 0.20 | 0.30 × 0.30 × 0.25 | 0.40 × 0.30 × 0.15 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | C2/c | P21/n | P21 |
| a/Å | 4.104(3) | 17.669(3) | 8.179(2) | 4.041(1) |
| b/Å | 5.427(6) | 8.058(3) | 8.826(3) | 5.641(3) |
| c/Å | 26.816(2) | 9.354(3) | 17.338(2) | 21.062(4) |
| β/° | 94.07(6) | 115.76(2) | 94.15(1) | 93.16(2) |
| V/Å3 | 595.8(8) | 1199.5(5) | 1248.2(5) | 479.3(3) |
| Z | 2 | 4 | 4 | 2 |
| d calcd/g cm−3 | 1.306 | 1.297 | 1.246 | 1.248 |
| μ(Cu-Kα)/cm−1 | 6.90 | |||
| μ(Mo-Kα)/cm−1 | 0.79 | 0.76 | 0.78 | |
| F(000) | 244.00 | 488.00 | 488.00 | 188.00 |
| No. of reflections, m with (I > 2σ(I)) | 642 | 773 | 1719 | 911 |
| No. of variables, n | 103 | 107 | 204 | 160 |
|
Ra |
0.042 | 0.060 | 0.050 | 0.040 |
|
Rwb |
0.049 | 0.066 | 0.049 | 0.042 |
|
GOFc |
3.32 | 1.51 | 1.69 | 1.59 |
| Δρmax/e Å−3 | 0.14 | 0.18 | 0.16 | 0.10 |
Full crystallographic details (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre (CCDC).
CCDC reference number 188/200.
Results and discussion
Upon heating 1a crystals (obtained from acetone–benzene mixtures) turned black above ca. 120 °C and did not melt beyond 300 °C. When recrystallized from pure acetone, colouration of 1b crystals was observed to begin at ca. 160 °C, and the black crystals melted at 210 °C. The melting point for crystals of 1 was therefore not determined because of the occurrence of polymerization upon heating.The crystal structure of 1 depends on the recrystallization solvents. Fig. 3 shows the crystal structure of 1a. The diacetylene groups are arranged regularly along the b axis. The regular arrangement along the b axis is formed by hydrogen bonding between neighbouring hydroxy groups.
 | ||
| Fig. 3 Crystal structure of 1a, recrystallized from acetone–benzene mixtures, viewed down the a axis. | ||
To define the arrangement of diacetylene monomers, the following parameters are used.6 They are intermolecular distance, R, between reacting carbons, stacking distance, d, between neighbouring monomers and the angle, ϕ, between diacetylene rod and stacking axis. In the crystal structure of 1a, R, d and ϕ are 3.92 Å, 5.43 Å and 44°, respectively; these parameters are in the range reported to be favorable for polymerization.6 Thus, the solid-state polymerization in this crystal is considered to proceed along the b axis. The 1a crystals polymerized so as to be insoluble in common organic solvents when irradiated with γ-rays or heated, and the colourless crystals became black, as expected from the crystal data. Fig. 4 shows the FTIR spectra of 1a, irradiated with γ rays at a dose of 2.0 MGy and heated at 100 °C for 25 h. The absorption intensity of the –CC–CC– band at 2240 cm−1 decreases and a new band appears at around 2190 cm−1, which is assigned to conjugated acetylene.8Fig. 5 shows the consumption of the butadiynyl groups in the radiation-induced polymerization of 1a. The consumption of the butadiynyl groups seems to be constant at below 50% above the irradiation dose of ca. 500 kGy. There was no appreciable change in the FTIR spectra at the high irradiation doses at least above 1.0 MGy. Therefore, the radiation-induced polymerization is considered to proceed by one of the diacetylene moieties. The arrangement of the remaining butadiynyl groups of the polymer should not be suitable for the further reactions of the pendants. Unfortunately, attempts at X-ray structure analysis of the samples in various stages of polymerization were unsuccessful because of the poor crystallinity of the samples, and the arrangement of the remaining butadiynyl groups of the polymer was not clarified.
 | ||
| Fig. 4 FTIR spectra of 1a (A) irradiated at a dose of 2.0 MGy and (B) heated at 100 °C for 25 h. | ||
 | ||
| Fig. 5 Consumption of the butadiynyl groups in the radiation-induced polymerization of 1a, obtained from acetone–benzene mixtures. | ||
Fig. 6 shows the consumption of the butadiynyl groups in the thermal polymerization at 100 and 150 °C. It is demonstrated that both of the diacetylene moieties participate in the thermal polymerization, resulting in the consumption of the butadiynyl groups above 50%. The FTIR spectral change around the 2240 and 2190 cm−1 bands at 100 °C is illustrated in Fig. 7. After polymerization by one of the butadiynyl groups, a rearrangement of the polymer may occur to promote the reactions of the pendants at the high temperatures.
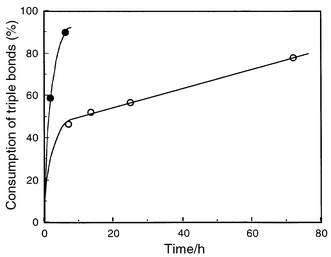 | ||
| Fig. 6 Consumption of the butadiynyl groups in the thermal polymerization of 1a, obtained from acetone–benzene mixtures, at (○) 100 and (•) 150 °C. | ||
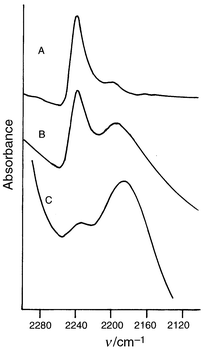 | ||
| Fig. 7 Change in the absorption intensities of the 2240 and 2190 cm−1 bands of the 1a crystals upon heating at 100 °C: the heating times are (A) 0, (B) 25 and (C) 72 h. | ||
Fig. 8 shows the crystal structure of 1b. It was found that the arrangement of the aromatic rings in the neighbouring monomer molecules is not parallel. The parameters R, d and ϕ are 6.08 Å, 9.35 Å and 24°, respectively, which indicate that the arrangement of this crystal is not suitable for the polymerization. The crystals did not polymerize upon γ-irradiation, as expected. 1b Crystals irradiated at a dose of 510 kGy were submitted to gel permeation chromatography. Even low molecular weight oligomers were not detected by the gel permeation chromatogram of the irradiated crystals. Neither did 1b polymerize upon heating at up to 150 °C.
 | ||
| Fig. 8 Crystal structure of 1b, recrystallized from acetone, viewed down the b axis. | ||
No crystals which polymerized upon γ-irradiation or heating were obtained from 2. The melting point of the crystals obtained from ethyl acetate was 135.2–136.0 °C. Fig. 9 shows the structure of the inert crystals. The parameters R, d and ϕ are 6.19 Å, 8.83 Å and 34°, respectively, which indicate that the arrangement of crystal 2 is unfavorable for polymerization.6
 | ||
| Fig. 9 Crystal structure of 2 viewed down the a axis. | ||
Figs. 10A and 10B show the crystal structure of 3, obtained from hexane–acetone mixtures. Considering the structure from the a axis projection of the crystal packing, the diacetylene groups are arranged along the b axis and their packing parameters R, d and ϕ are 4.02 Å, 5.64 Å and 42°, respectively (Fig. 10A). It was found that this crystal structure is quite similar to the crystal structure of 1a and the crystals of 3 take suitable packing structures for solid-state polymerization. Therefore, it is expected that this monomer is polymerized along the b axis. The regular arrangement along the b axis is also formed by hydrogen bonding between neighbouring hydroxy groups. On the other hand, from the crystal packing projected along the b axis, it was found that the ethynyl moieties are regularly arranged along the a axis and the shortest carbon–carbon distance of the neighbouring ethynyl moieties is 3.79 Å (Fig. 10B). On the basis of the molecular arrangement, the solid-state polymerization of 3 by the ethynyl moiety is expected to proceed, similarly to the case of 1,4-diethynylbenzene.1,18 Thus, with respect to the crystals of 3, the monomer arrangement is suitable for solid-state polymerization not only by the diacetylene moieties but also by the ethynyl groups. The distance between the reacting carbons is shorter for the ethynyl groups than for the diacetylene moieties. So polymerization is expected to be preferable for the ethynyl groups than for the diacetylene moieties. Irradiation of the crystals of 3 for a short time resulted in a poor crystallinity. This may be due to the scission of the hydrogen bonds between the neighbouring diacetylene moieties caused by the predominant bond formation between the ethynyl groups. Such a lowering of the crystallinity was not observed for the irradiated 1a crystals, where the crystal lattice is held by the diacetylene polymerization.
 | ||
| Fig. 10 Crystal structure of 3 viewed down the (A) a and (B) b axes. | ||
Crystals of 3 polymerize upon γ-irradiation but not upon heating. A decrease in absorption intensity at 2240 cm−1 was observed in the FTIR spectra of the irradiated crystals of 3, as well as the appearance of a new band at 2190 cm−1, similarly to the case of 1a. In addition, the absorption intensity of the –CC–H band at 3263 cm−1 decreased upon γ-irradiation. These results indicate that both of the ethynyl and butadiynyl groups participate in the polymerization, as expected from the crystallographic data. Fig. 11 shows the consumption of the ethynyl and butadiynyl groups in the radiation-induced polymerization of 3. The consumption of the ethynyl groups is larger than that of the butadiynyl groups. Furthermore, the consumption of the butadiynyl groups of 3 is smaller than that of 1a. These results can be interpreted in terms of the long distance between the reacting carbons of the butadiynyl groups of 3, 4.02 Å, compared with that of 1a, 3.92 Å. Polymerization by the butadiynyl groups may have an induction period (Fig. 11), although there may be some errors in the low yields in the early stage of the polymerization. On the basis of the molecular arrangement of the crystals, the two propagations by the ethynyl and butadiynyl groups of 3 are considered to proceed along the different axes of the crystals.
 | ||
| Fig. 11 Consumption of the (○) ethynyl and (•) butadiynyl groups in the radiation-induced polymerization of 3. | ||
The relatively long distance between the reacting carbons of the butadiynyl group of 3 may be responsible for its inertness toward heating. The ethynyl groups may also be inert toward heating. In our previous study,1,2 1,4-diethynylbenzene did not polymerize upon heating below the melting point, 94–95 °C, whereas it polymerized upon irradiation with γ-rays1 and UV light.18 The ethynyl groups attached to aromatic rings are considered to be inert in the thermal polymerization in crystalline states, since the melting points of various aromatic acetylenes have been determined.18
References
- M. Hagihara, Y. Yamamoto, S. Takahashi and K. Hayashi, Radiat. Phys. Chem., 1986, 28, 165 CAS.
- Y. Kai, A. Yamamoto, D. Xu, N. Kasai, M. Hagihara, Y. Yamamoto, S. Takahashi and K. Hayashi, Makromol. Chem., 1987, 188, 3047 Search PubMed.
- Y. Yamamoto, T. Ueda, N. Kanehisa, K. Miyawaki, Y. Takai, S. Tagawa, M. Sawada and Y. Kai, J. Chem. Soc., Perkin Trans. 2, 1997, 734 Search PubMed.
- H. Basseler, Adv. Polym. Sci., 1984, 63, 1 Search PubMed.
- H. Sixl, Adv. Polym. Sci., 1984, 63, 49 Search PubMed.
- V. Enkelmann, Adv. Polym. Sci., 1984, 63, 91 Search PubMed.
- S. Okada, H. Matsuda, A. Masaki, H. Nakanishi and K. Hayamizu, Chem. Lett., 1997, 1105.
- S. Okada, H. Matsuda, H. Nakanishi and M. Kato, Mol. Cryst. Liq. Cryst., 1990, 189, 57 Search PubMed.
- S. Okada, K. Hayamizu, H. Matsuda, A. Masaki and H. Nakanishi, Bull. Chem. Soc. Jpn., 1991, 64, 857 CAS.
- S. Okada, K. Hayamizu, H. Matsuda, A. Masaki, N. Minami and H. Nakanishi, Macromolecules, 1994, 27, 6259 CrossRef CAS.
- K. Hayamizu, S. Okada, T. Doi, H. Kawanami, N. Kikuchi, H. Matsuda and H. Nakanishi, Bull. Chem. Soc. Jpn., 1995, 68, 791 CAS.
- S. Nezu and J. B. Lando, J. Polym. Sci.: Part A: Polym. Chem., 1995, 33, 2455 Search PubMed.
- W. S. Price, N. Kikuchi, H. Matsuda, K. Hayamizu, S. Okada and H. Nakanishi, Macromolecules, 1995, 28, 5363 CrossRef CAS.
- H. Matsuzawa, S. Okada, H. Matsuda and H. Nakanishi, Chem. Lett., 1997, 1105.
- L. Brandsma , Preparative Acetylenic Chemistry, Elsevier, Amsterdam, 1988, ch. 10, p. 212. Search PubMed.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 CrossRef CAS.
- T. Ando, S. Shioi and M. Nakagawa, Bull. Chem. Soc. Jpn., 1972, 45, 2611 CAS.
- O. Rohde and G. Wegner, Makromol. Chem., 1978, 179, 2013 Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |