Role of the solvent in optical resolution of trans-chrysanthemic acid via diastereomeric salt formation with (1R,2R)-1-(4-nitrophenyl)-2-dimethylaminopropane-1,3-diol
Éva Kozsda-Kovácsa, György Miklós Keserüb, György Miklós Keserü*c, Zsolt Böcskeic, Ildikó Szilágyic, Kálmán Simonc, Béla Bertóka and Elemér Fogassy*d
aAGRO-CHEMIE Ltd., P.O. Box 49, H-1780, Budapest, Hungary
bDepartment of Chemical Information Technology, Technical University of Budapest, Szt. Gellért tér 4, H-1111, Budapest, Hungary
cCHINOIN Pharmaceuticals, Tó u. 1-4, H-1134, Budapest, Hungary
dDepartment of Organic Chemical Technology, Technical University of Budapest, Mûegyetem rkp. 3, H-1111, Budapest, Hungary
First published on UnassignedUnassigned23rd December 1999
Abstract
Optical resolution of trans-chrysanthemic acid via diastereomeric salt formation with (1R,2R)-1-(4-nitrophenyl)-2-dimethylaminopropane-1,3-diol (DMAD) has been studied in different solvents. Ether type solvents containing MeOH were found to be preferred. The role of MeOH was interpreted on the basis of powder and single crystal X-ray diffraction and DSC/TG measurements. We found that MeOH was incorporated into the crystals of the less soluble diastereomer salt of the 1R acid with DMAD in a non-stochiometric amount and postulated to promote nucleation and crystal growth.
Introduction
Optical resolution of racemic acids and bases via diastereomeric salt formation is the most popular and convenient method for the separation of enantiomers.1 Although this is also the most preferred technology on an industrial scale, the molecular basis of these processes has rarely been clarified. Since optical resolutions usually depend on many correlated parameters, investigations at the molecular level might be helpful in laboratory and pilot scale optimisation and may promote the development of an effective industrial scale process. Finding the best resolving agent and optimal conditions is a challenging task, which is still largely based on trial-and-error. Structural data obtained by the systematic study of diastereomeric salt formations are useful for the rationalisation of optical resolution and help in the experimental design of the most important parameters.2Cyclopropanecarboxylic acids (permethrinic and chrysanthemic acids, Scheme 1) are used as a main building block in the synthesis of pyrethroid insecticides.3 Although there are a number of products marketed as a racemic mixture, the minimum risk concept of environmental protection agencies makes the application of pure enantiomers highly desirable. Optically active pyrethroids such as Bioallethrin and Deltamethrin (Scheme 1) are widely used in household and agrochemical protection. The optical resolution of permethrinic acid is used to prepare optically active analogues of Permethrin, while the optically active (+)-trans-chrysanthemic acid is the starting material for the preparation of Bioallethrin and its analogues. An efficient resolution of permethrinic acids on the industrial scale has been published by our laboratory.4 Optimal conditions were rationalised by a number of structural studies on the diastereomeric salts.5 Here, we report the molecular basis of the resolution of chrysanthemic acids. Although there are a number of related processes reported earlier,6 this is the first time that this chiral discrimination has been investigated at a molecular level.
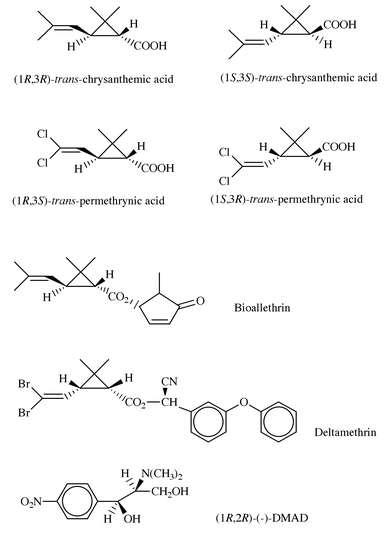 | ||
| Scheme 1 | ||
Results and discussion
Optical resolution
Optical resolution of chrysanthemic acid was originally developed by Roussel-Uclaf![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 6 utilising (1R,2R)-1-(4-nitrophenyl)-2-dimethylaminopropane-1,3-diol (DMAD) as a chiral base in diisopropyl ether–methanol (6∶1). Considering the high chemical and optical yield of the process, DMAD a readily available intermediate of the well known antibiotic chloramphenicol, seems to be an optimal resolving agent. Optimising the resolution process we concluded that the role of the solvent should be clarified. In this study we report a systematic investigation on the resolution of trans-chrysanthemic acid using different solvent systems.
6 utilising (1R,2R)-1-(4-nitrophenyl)-2-dimethylaminopropane-1,3-diol (DMAD) as a chiral base in diisopropyl ether–methanol (6∶1). Considering the high chemical and optical yield of the process, DMAD a readily available intermediate of the well known antibiotic chloramphenicol, seems to be an optimal resolving agent. Optimising the resolution process we concluded that the role of the solvent should be clarified. In this study we report a systematic investigation on the resolution of trans-chrysanthemic acid using different solvent systems.Chrysanthemic acid was resolved in a number of solvents and solvent mixtures and the optical purity and the resolving efficiency of each system was determined (Table 1). The latter was quantified by the parameter S7 which is the product of the chemical yield and the optical purity. We found that an acceptable resolving efficiency and even crystallisation could only be achieved in the presence of MeOH. Resolution in pure methanol gave, however, a low chemical yield. Therefore several solvent mixtures containing a variable amount of MeOH have been tested. Although we found the optical purity with halogenated hydrocarbons to be acceptable, the resolving efficiency of these systems was low. Resolutions in mixtures with hydrocarbons or a second polar component also resulted in low chemical yields. The application of ether-type solvents [di-n-butyl ether, THF, methyl tert-butyl ether (MTBE) and diethyl ether] in mixtures with MeOH, however, gave high optical purity. The application of a 6∶1 mixture of di-n-butyl ether and MeOH resulted in a higher chemical yield but lower optical purity than those of the original process using diisopropyl ether–methanol (6∶1).6 Although the optical purity of trans-crysanthemic acid can be increased using a 3∶1 mixture of THF and MeOH, the chemical yield of this resolution was found to be extremely low. Both the chemical yield and the optical purity of the product could be enhanced using a 1∶1 mixture of MTBE and MeOH. Best results were obtained using a 1∶1 mixture of diethyl ether with MeOH which yields the highest resolving capability (S = 0.86).
| Solvent | Yield (%) | Optical purity (%) | Resolving efficiency (S) |
|---|---|---|---|
| a No crystallisation observed. | |||
| Diisopropyl ether–MeOH (6∶1) | 84.4 | 96.7 | 0.82 |
| n-Hexane | —a | ||
| EtOAc | —a | ||
| iso-Butanol | —a | ||
| Methyl tert-butyl ether (MTBE) | 62.0 | 18.4 | 0.11 |
| MeOH | 52.2 | 93.1 | 0.48 |
| Water–MeOH (1∶1) | 67.6 | 80.5 | 0.54 |
| Cyclohexane–MeOH (1∶1) | 80.0 | 56.3 | 0.45 |
| CH2Cl2–MeOH (1∶1) | 40.0 | 82.7 | 0.33 |
| 1,2-Dichloroethane–MeOH (1∶1) | 38.0 | 72.1 | 0.27 |
| CHCl3–MeOH (1∶1) | 48.0 | 69.1 | 0.33 |
| CCl4–MeOH (1∶1) | 26.0 | 89.0 | 0.23 |
| Butan-2-one–H2O (1∶1) | 46.0 | 61.4 | 0.28 |
| MTBE–MeOH (1∶1) | 70.7 | 97.1 | 0.69 |
| Di-n-butyl ether–MeOH (6∶1) | 99.2 | 83.9 | 0.83 |
| Diethyl ether–MeOH (1∶1) | 86.5 | 99.3 | 0.86 |
| THF–MeOH (3∶1) | 15.5 | 91.4 | 0.14 |
Since the presence of MeOH seems to be a condition for successful resolution, we speculated that crystals of the diastereomeric salt might contain MeOH. The molecular structure of the diastereomeric salt 1 obtained from the resolution in MTBE–MeOH and pure salts prepared from trans-(+)- and -(−)-chrysanthemic acids and DMAD in EtOAc (2 and 3, respectively) were investigated by X-ray crystallography, powder diffraction analysis and DSC–TG measurements.
Powder diffraction studies
Powder diffraction analysis of 1, 2 and 3 revealed that each of the diastereomeric salts have different crystal forms. Salts containing the 1R-trans enantiomer (1 and 2) are pseudo-polymorphic and differ significantly from 3. Since MeOH was used only for the preparation of 1 we concluded that this pseudo-polymorphic relationship might indicate the involvement of MeOH in the crystal structure of 1. Clear differences between the powder diffraction data of 1 and 3, however, can be attributed to the presence of enantiomeric acids.X-Ray diffraction studies
Investigating the role of MeOH in the optical resolution process, the crystal structures of 1, 2 and 3 were determined [Fig. 1(a), (b) and (c), respectively]. Crystal data and details of structure refinement are collected in Table 2. Our objectives were (i) to study the role of MeOH in the resolution process; (ii) to understand the structural background of stability differences as well as (iii) to find out more details about the structural determinants of the chiral discrimination process.| RTCADAM 1 | RTCADA 2 | STCADA 3 | |
|---|---|---|---|
| Empirical formula | C22H36N2O7 | C21H32N2O6 | C21H33N2O6 |
| M | 440.53 | 408.49 | 408.49 |
| T/K | 293(2) | 293(2) | 293(2) |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | P212121 | P21 | P212121 |
| Unit cell dimensions | |||
| a/Å | 13.21(2) | 9.92(1) | 13.288(4) |
| b/Å | 26.59(2) | 7.66(1) | 27.298(5) |
| c/Å | 7.22(2) | 15.57(1) | 6.235(6) |
| β/° | — | 107.0(1) | — |
| V/Å3 | 2535(8) | 1132(2) | 2262(2) |
| Z | 4 | 2 | 4 |
| μ/mm−1 | 0.706 | 0.088 | 0.721 |
| Independent reflections | 2843 | 1269 | 2658 |
| Data/restraints/parameters | 2819/0/287 | 1269/179/268 | 2656/0/271 |
| Goodness-of-fit on F2 | 1.094 | 1.017 | 1.064 |
| Final R indices [I > 2σ(I)] | R1 = 0.0628, wR2 = 0.1021 | R1 = 0.0989, wR2 = 0.2649 | R1 = 0.0562, wR2 = 0.1502 |
| R indices (all data) | R1 = 0.3433, wR2 = 0.2161 | R1 = 0.1323, wR2 = 0.3124 | R1 = 0.1175, wR2 = 0.1918 |
| Largest diff. peak, hole/e Å−3 | 0.237, −0.214 | 0.556, −0.408 | 0.222, −0.198 |
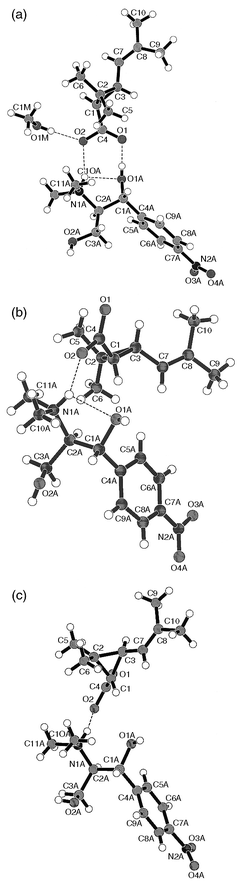 | ||
| Fig. 1 Structure of the ion pairs of 1 (a), 2 (b) and 3 (c) in their crystals. | ||
All three crystal structures are in accord with the expected conformations (trans-chrysanthemic acid). The absolute configuration of compounds 1, 2 and 3 could not be determined by X-ray crystallography and were assigned on the basis of the known configuration of the starting materials. Bond lengths and angles are in the expected ranges in all three structures.
The structure of a crystal formed in a resolution experiment (1, code: RTCADAM) contains one molecule of MeOH. The thermal motion parameters of the atoms of the MeOH molecule are twice as high on average compared to those of the other atoms which may point to partial occupation of the methanol position. The structure containing MeOH (1) is principally different from that of the same salt without any crystal solvent (2, code RTCADA). Crystals of 1 and 2 belong to different space groups, with RTCADAM 1 crystallising in the higher symmetry orthorhombic system. Interestingly, the crystal structure containing the 1S acid (3, code STCADA) has very similar cell dimensions and identical space group to RTCADAM 1, the only structure crystallised with MeOH.
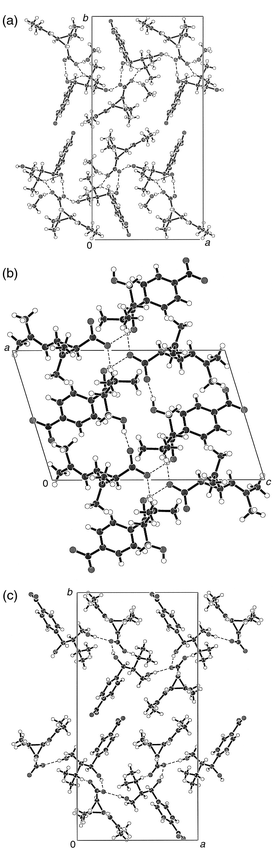 | ||
| Fig. 2 Crystal packing diagrams of 1 (a), 2 (b) and 3 (c). Hydrogen bonds are shown with dashed lines, heteroatoms with different shadings. | ||
The differences in the stability of the three compounds may be explained on the basis of the hydrogen bond networks present in the structures (Table 3). Melting point differences and solubility studies (Table 4) revealed that the diastereomeric salt containing DMAD and 1Rtrans-acid is more stable than that formed with 1S acid. This is also demonstrated by the bridgehead atom distances of the hydrogen bonds. First we compared the structures of the two diastereomeric salts which do not contain the solvent MeOH (2 and 3). The salt bridges between the ammonium NH and one of the carboxylate oxygens and one of the hydrogen bonds formed by the hydroxy groups of DMAD have nearly equal strengths. However, the second OH⋯O type hydrogen bond appears to be significantly stronger in 2. In the methanol containing crystal (1) there are four types of intermolecular hydrogen bonds since the MeOH molecule participates in the hydrogen bonding network. Two of them are about as strong as those in the crystal of 2, while the third is weaker. The fourth one, which is formed by methanol may, however, provide extra stability for the MeOH containing crystals of 1.
| Compound | Hydrogen bond | D⋯A/Å | H⋯A/Å | D–H⋯A/° |
|---|---|---|---|---|
| 1 | OM1–H10M⋯O1 | 2.645(17) | 1.86(2) | 171(1) |
| O1A–H1A⋯O1 | 2.619(13) | 1.81(2) | 168(10) | |
| O2A–H2A⋯O1 | 2.694(14) | 1.92(4) | 158(11) | |
| N1A–H1A1⋯O2 | 2.780(15) | 1.97(1) | 147(1) | |
| N1A–H1A1⋯O1A | 2.806(15) | 2.27(1) | 118(1) | |
| 2 | O1A–H1AO⋯O2 | 2.642(11) | 1.85(5) | 162(14) |
| O2A–H2A⋯O1 | 2.693(14) | 2.00(21) | 141(31) | |
| N1A–H1AN⋯O2 | 2.668(12) | 1.83(1) | 152(1) | |
| N1A–H1AN⋯O1A | 2.687(13) | 2.25(1) | 109(1) | |
| 3 | O2A–H2AO⋯O1 | 2.702(5) | 1.88(1) | 176(4) |
| O1A–H1AO⋯O1 | 2.711(5) | 1.91(6) | 166(2) | |
| N1A–H1AN⋯O2 | 2.656(6) | 1.83(1) | 150(1) | |
| N1A–H1AN⋯O1A | 2.746(6) | 2.42(1) | 101(1) |
A more detailed view of the hydrogen bonding topology reveals further interesting structural features. All intermolecular H-bonds cluster around the carboxylate groups in all three structures. In 1 all lone pairs of the carboxylate oxygens have H-bond donor partners. O2 accepts H-bonds from the MeOH molecule and from the protonated nitrogen of DMAD. O1 accepts H-bonds from the non-identical hydroxy groups of two cations. In contrast, however, there are only three hydrogen bonds around the carboxylate groups in 2 and 3. These hydrogen bonds originate from three different cations in the vicinity of the carboxylate in both 2 and 3. It is interesting that the protonated nitrogen of DMAD donates an H-bond to the carboxylate in 2 and 3 from the very position where the OH group of MeOH is located in 1. On the other hand in 2, at the position which in 1 is occupied by the protonated nitrogen, we found a hydroxy group of the base. All three structures contain several relatively short C⋯O distances in about equal number. These C–H⋯O hydrogen bonds are characterised with bridgehead atom distances of between 3.0 and 3.5 Å. These include multipoint type interactions between methyl groups,8 aromatic C–H as well as oxo, alcoholic hydroxy and nitro type oxygens.
On the basis of powder diffraction data we propose that the presence of MeOH is responsible for the pseudo-polymorphism observed between 1 and 2. X-Ray diffraction analysis of these compounds revealed the presence of a MeOH molecule in the crystal structure of 1. Excellent agreement between the experimental powder diffraction diagrams and those calculated on the basis of single crystal measurements confirms our first intuition, i.e. that crystals of the less soluble diastereomeric salt (1) might contain MeOH.
DSC analysis
The different stabilities of 1, 2 and 3 have been interpreted on a molecular basis. Thermal consequences of structural differences were thus investigated by TG and DSC. DSC data obtained for 1, 2 and 3 are in accordance with the higher stability of 1 and 2 over 3. Thus melting points for 1 and 2 (Table 4) are higher than that of 3. The slightly higher melting point of 1 compared to 2 also suggested that 1 was more stable and underlined the role of MeOH in the chiral discrimination process. Comparing DSC curves of 1, 2 and 3 we found that 1 underwent polymorphic rearrangements at 95 and 97.7 °C which could not be detected in 2 or 3. Note, however, that DTG measurement on 1 detected only 0.25 mol equivalents of MeOH in 1. In contrast, DSC–TG analysis of 1 recrystallised from pure MeOH showed no polymorphic rearrangement while DTG indicated the presence of 1.03 mol equivalents of MeOH in the sample.Interpretation of results
The process of crystallisation can be divided into two distinct phases: nucleation and crystal growth. Based on our circumstantial evidence we suggest the most stable diastereomeric salt 1 incorporating MeOH is involved in nucleation and in the early stage of crystal growth. Since crystallisation begins with solute–solvent aggregates that contain solute–solute, solute–solvent and solvent–solvent interactions, this role of MeOH is not unexpected. A recent analysis of crystal structures8 deposited at the Cambridge Structural Database revealed that MeOH is the solvent most frequently found in molecular crystals.As has been pointed out by Nangia and Desiraju,8 the entropic gain in eliminating solvent molecules from the preformed aggregates as well as the enthalpic gain associated with the formation of stable solute species provides the driving force of nucleation to yield unsolvated organic crystals. On the other hand hydrogen bonds formed between the solvent and the solute—which is typical for acids and alcohols—make the extrusion of the solvent unfavourable. Solvated crystals of these compounds usually include MeOH or EtOH since they have a very good donor and a moderate acceptor group. The inclusion of MeOH was evident in 1 and therefore we propose that MeOH remained an integral part of the nucleating crystal. This phase of crystallisation is thought to determine the resolving efficiency, which correlates with our observation that high resolving capability could only be achieved in the presence of MeOH. Owing to the co-crystallisation of MeOH the space around the symmetry element in the crystal is conveniently filled since the crystallising ion pairs are prevented from occupying the crystal symmetry elements owing to steric factors.9 Therefore we suggest that the role of the MeOH in the crystal lattice is to ensure close packing which would not be achievable by packing of the low energy conformation of the given diastereomer alone. Since nucleation is also the rate-limiting process of crystallisation, we propose that MeOH promotes the crystallisation of the less soluble diastereomeric salt which crystallises without MeOH (2) in the final stage.
Summary
The molecular basis of the optical resolution of trans-chrysanthemic acid has been investigated. Solvent systems containing MeOH were found to be optimal for chiral discrimination. The crucial role of MeOH was rationalised by powder diffraction analysis, X-ray diffraction studies, and DSC–TG measurements. We found that MeOH detected in the diastereomeric salt obtained from the resolution process has a characteristic effect on hydrogen bonding within the unit cell. The increased stability of the DMAD/1R-trans acid salt 2 can be interpreted by a more extended H-bonding network as compared to that found in 3. Additional H-bonds as a consequence of the presence of MeOH in 1 are responsible for the effective chiral discrimination observed in resolution experiments using mixtures of MeOH and ether-type solvents. DSC–TG measurements revealed a non-stoichiometric amount of MeOH incorporated in crystals of 1. Since 1 was produced in the resolution process, we propose that MeOH promoted the crystallisation of the diastereomeric salt of the 1R acid with DMAD during the nucleation phase and at the very beginning of crystal growth.Experimental
Thermal data were recorded on a SETARAM TGDSC111 apparatus while optical rotation measurements were measured using a Perkin-Elmer 241 polarimeter.Optical resolution of trans-chrysanthemic acid
To 16.8 g (0.1 mol) of racemic trans-chrysanthemic acid and 24.0 g (0.1 mol) of DMAD was added a 1∶1 mixture of diethyl ether and methanol (75 ml) and the mixture heated to 43 °C to allow dissolution. On allowing to cool for 10 min crystallisation commenced at 33 °C. After 1 h at 0–5 °C the crystals were filtered off and washed with the cold solvent mixture. Then 15 ml of 5 M aq. HCl, 15 ml of dichloroethane and 15 ml of water were added. After 1 h of stirring the organic layer was separated, dried over anhydrous MgSO4 and evaporated. The resulting 1R-(+)-enantiomer was obtained, 6.7 g (86.5%), [α]20D = 26.5 (c 1, CHCl3).X-Ray analyses
Powder diffraction analyses were performed on a Philips PW3710 diffractometer utilising Cu-Kα (λ = 1.5418 Å) radiation (λ1 and λ2 were 1.54060 and 1.54439 Å, respectively, with a λ1∶λ2 ratio of 2∶1). A step size of 0.02° in the 2θ range 3–30° was applied in all measurements.Single crystals of 1 were grown from a MeOH–methyl tert-butyl ether mixture, while those of 2 and 3 were grown from EtOAc. Drawings with numbering schemes of all three ion pairs are shown on Fig. 1(a), (b) and (c), respectively. Crystal data for 2 were collected on a R-AXIS II imaging plate equipped with a Rigaku RU-200 generator, while those for 1 and 3 were collected on a Rigaku AFC6S diffractometer. Graphite monochromated Cu-Kα radiation was used in the latter two cases, while Mo-Kα was used on the imaging plate. The structures were solved using the TEXSAN package10 and refined with SHELXL-93.11 Because of the low data/parameter ratio in the refinement of 2, planarity and isotropic restraints were included. Crystal data and structure refinements are detailed in Table 2.
CCDC reference no 188/196. See http://www.rsc.org/suppdata/p2/a9/a904682h/ for crystallographic files in .cif format.
Acknowledgements
We are grateful for the financial support of the OTKA Foundation (grant No. F 019261).References
- J. Jacques , A. Collet and H. Wilen , Enantiomers, Racemates, and Resolutions, Krieger Publishing Company, Malabar, FL, 1994. Search PubMed.
- P. M. C. Brianso, Acta Crystallogr., Sect. C, 1976, 32, 3040 CrossRef; R. O. Gould and M. D. Walkinshaw, J. Am. Chem. Soc., 1984, 106, 7840 CrossRef CAS; E. Fogassy, M. Ács, F. Faigl, K. Simon, J. Rohonczy and Z. Ecseri, J. Chem. Soc., Perkin Trans. 2, 1986, 1881 RSC; M. Czugler, I. Csöregh, A. Kálmán, F. Faigl and M. Ács, J. Mol. Struct., 1989, 196, 157 CrossRef CAS; M. Ács, E. Nowotny-Bregger, K. Simon and G. Argay, J. Chem. Soc., Perkin Trans. 2, 1992, 2011 RSC; K. Hatano, T. Takeda and I. Saito, J. Chem. Soc., Perkin Trans. 2, 1994, 579 RSC; K. Kinbara, K. Sakai, Y. Hashimoto, H. Nohira and K. Saigo, J. Chem. Soc., Perkin Trans. 2, 1996, 2615 RSC; K. Kinbara, Y. Kobayashi and K. Saigo, J. Chem. Soc., Perkin Trans. 2, 1998, 1765 RSC.
- M. Elliott , in Synthetic Pyrethroids, ACS Symp. Ser. 42, ed. M. Elliott, Washington D.C., 1977, p. 8. Search PubMed.
- B. Bertók, I. Czudor, I. Székely, L. Pap, L. Csíz and P. Forgó, J. Environ. Sci. Health B, 1996, 31, 495.
- E. Fogassy, F. Faigl, M. Ács, K. Simon, É. Kozsda, B. Podányi, M. Czugler and G. Reck, J. Chem. Soc., Perkin Trans. 2, 1988, 1385 RSC; F. Faigl, K. Simon, A. Lopata, É. Kozsda, R. Hargitai, M. Czugler, M. Ács and E. Fogassy, J. Chem. Soc., Perkin Trans. 2, 1990, 57 RSC.
- Roussel. Uclaf, 1980, BP 2 039 894; Search PubMed; Stauffer Chemical Company, 1982, HU 192 763; Search PubMed; Roussel. Uclaf, 1979, HU 181 948; Search PubMed; Stauffer Chemical Company, 1983, EP 68 736; Search PubMed; Sumitomo Corp., 1988, Jpn P 63 35 540; Search PubMed; Kuraray Co. Ltd., 1991, Jpn. P 03 74 347; Search PubMed; Hiroyuki Co., 1986, Jpn. P 61 172 853; Search PubMed; Roussel. Uclaf, 1981, US. Pat., 4 257 976; Search PubMed; Sumitomo Corp., 1992, EP No. 508 307. Search PubMed.
- E. Fogassy, A. Lopata, F. Faigl, F. Darvas, M. Ács and L. Tõke, Tetrahedron Lett., 1980, 21, 647 CrossRef CAS.
- A. Nangia and G. R. Desiraju, Chem. Commun., 1999, 605 RSC.
- P. van der Sluis and J. Kroon, J. Cryst. Growth, 1989, 97, 645 CrossRef CAS.
- TEXSAN, Single Crystal Structure Analysis Package, Molecular Structure Corporation, The Woodlands, TX 77381, 1992. Search PubMed.
- G. M. Sheldrick , SHELXL-93, Program for the Refinement of Crystal Structures, University of Göttingen, 1993. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |