C
3-symmetric ferrichrome-mimicking Fe3+-complexes containing the 1-hydroxypyrimidinone Fe3+-binding moieties based on α-cyclodextrin: helicities in solvent environments
Received
(in Cambridge, UK)
24th May 1999
, Accepted 26th October 1999
First published on 14th January 2000
Abstract
Two homologous C3-symmetric ferrichrome mimics, La and Lb, equipped with three 1-hydroxypyrimidinone terminals for receiving an Fe3+ ion have been designed and synthesized; the side chains of La and Lb, which include dimethylene and pentamethylene spacers, respectively, are anchored to the narrower rim of the α-cyclodextrin framework through an amide linkage. The 1∶1 Fe3+-complexes of La and Lb provide strong circular dichroic exciton coupling of negative and positive signs at 480 nm, showing Δ- and Λ-helices, respectively, in polar aprotic solvents such as dimethylformamide and dimethyl sulfoxide. The magnitude of the signals increases with the increasing hydrogen-bond accepting ability of the solvent. It is inferred, therefore, that hydrogen-bonding interactions of the linker amide N–H groups with the solvent play a primary role in inducing a helicity within aprotic solvents. The mechanisms of the helicity inductions are discussed.
Meanwhile, the signals for La are still negative, but weak in alcohols and water. However, the signal sign for Lb having the relatively flexible spacers depends on the nature of alcohols, and no CD signal was observed in water.
Naturally occurring siderophores such as ferrichrome and enterobactin are tripodal chiral Fe3+-specific chelating agents and their production is required for solubilization and transportation of Fe3+ ions in microorganisms![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 1 in which the Fe3+ ion is embedded in the octahedral cavity of three bidentate ligands on the side-chains. Iron chelation is also important in therapeutic treatments of a human genetic disease, Cooley’s anemia.2 Many synthetic or natural siderophores have been tested for the therapic treatments, and desferrioxamine B, a simple linear ferrichrome-mimicking compound, has been developed for practical utility.
1 in which the Fe3+ ion is embedded in the octahedral cavity of three bidentate ligands on the side-chains. Iron chelation is also important in therapeutic treatments of a human genetic disease, Cooley’s anemia.2 Many synthetic or natural siderophores have been tested for the therapic treatments, and desferrioxamine B, a simple linear ferrichrome-mimicking compound, has been developed for practical utility.
Among various ferrichrome mimics, tripodal types of compounds in particular have attracted our attention thus far, because covalently linking the ligand chains in a C3-symmetric way to a common anchor would reduce the number of possible complexed species and enable us to unequivocally elucidate the most feasible conformation of the complex.3 Recent publications on tripodal Fe3+ complexes4 prompted us to design another type of tripodal complex and to implicate the mechanism of the induced helicity; as part of our continuing research we have recently reported C3-symmetric tripodal mimics constructed on the narrower face of the rigid α-cyclodextrin framework (α-CyD) instead of a cyclic lactone ring in the natural ferrichrome, and shown that the linking of three monohydroxamate side-chains to the α-CyD results in a twisting of the octahedral chromophore around the Fe3+ ion to preferentially produce a Δ-cis,cis complex with a right handed helicity (observed CD amplitude/mdeg: −26 (H2O) < −16 (MeOH) < −8 (acetonitrile) < −2 (dimethyl sulfoxide, no complexation) < −1 (pyridine, no complexation)),5 and the extent of the right-handedness decreases with decreasing solvent polarity and/or hydrogen bond donating ability of the solvent. The chiral cyclodextrin skeleton is an origin of induced helical chirality around the Fe3+ ion in an octahedral cavity. Meanwhile, the ligand having bis-hydroxamate side chains (Lc, Fig. 1), anticipated to form a triply helical binuclear complex, was found to provide a Δ-cis,cis complex in water. However, complexation in rather basic aprotic solvents such as pyridine and acetone led to the Λ-cis,cis complex with absolutely opposite helicity (observed CD amplitude/mdeg: −10 (H2O) < −3 (MeOH) < 0 (dimethyl sulfoxide, no complexation) < +5 (acetonitrile) < +8 (acetone) < +15 (pyridine)).6 We have suggested, accordingly, the primary importance of steric repulsion between the hydroxamate carbonyl and linker carbonyl groups and intermolecular hydrogen-bonding with the solvent in enhancing the Λ-helicity.
 |
| | Fig. 1 Chemical structures of ligands.
| |
To provide extensive systematic investigations for these fundamentally interesting concepts, we will report here the synthesis and physico-chemical properties of C3-symmetric ferrichrome surrogates (La and Lb in Fig. 1) that are assembled from an α-cyclodextrin as a tripodal anchor and three pendant side chains with 1-hydroxypyrimidinone terminals as an iron binding site.7 We have now found that the effects of solvent environments on helical chirality substantially differ for the hydroxamate and 1-hydroxypyrimidinone complexes. Helical chirality has currently been a central feature of chemical systems which are involved in a wide variety of biologically and technologically important processes.8
Experimental
Materials
Solvents such as alcohols, tetrahydrofuran (THF), acetonitrile (MeCN), acetone (Me2CO), formamide (HCONH2), dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were all distilled after drying before use. Fe2(NO3)2·8H2O was purchased from Tokyo Kasei Co. (Tokyo, Japan) and used as received. Ligands La and Lb were prepared according to Scheme 1. The preparation of the key intermediate 4a, the very same compound, had been achieved by Katoh et al.9 Therefore, the synthetic procedures for 2a, 3a, and 4a have been omitted here.
 |
| | Scheme 1 | |
4-[5-(Methoxycarbonyl)pentylamino]-1-(benzyloxy)pyrimidin-2(1H![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )-one (2b)..
1-Benzyloxy-4-(1,2,4-triazol-1-yl)pyrimidin-2(1H)-one10 (1, 960 mg, 3.57 mmol) was treated with the HCl salt of methyl 6-aminohexanoate (960 mg, 5.3 mmol) and triethylamine (0.73 mL, 5.3 mmol) in 30 mL of dried THF and stirred at room temperature for 14 h. After addition of 15 mL of water, the mixture was extracted five times with CHCl3.
)-one (2b)..
1-Benzyloxy-4-(1,2,4-triazol-1-yl)pyrimidin-2(1H)-one10 (1, 960 mg, 3.57 mmol) was treated with the HCl salt of methyl 6-aminohexanoate (960 mg, 5.3 mmol) and triethylamine (0.73 mL, 5.3 mmol) in 30 mL of dried THF and stirred at room temperature for 14 h. After addition of 15 mL of water, the mixture was extracted five times with CHCl3.
Evaporation of the solvent and recrystallization of the residue from EtOH gave 2b (640 mg, 1.86 mmol, 52%): mp 144–145 °C; IR(KBr) 3025, 1672, 1616, 746, 700 cm−1; Anal. Found: C, 62.43; H, 6.88; N, 12.11%. Calcd for C18H23N3O4: C, 62.59; H, 6.92; N, 12.17%; 1H NMR(CDCl3) δ 1.37–1.39 (m, 2H, 3-CH2), 1.60–1.66 (m, 4H, 2,4-CH2), 2.32 (t, J = 7.4 Hz, 2H, CH2CO), 3.22–3.28 (m, 2H, NHCH2), 3.67 (s, 3H, CH3O), 5.07 (s, 2H, CH2Ph), 5.53 (d, J = 7.6 Hz, 1H, N–CH![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH), 7.40–7.45 (m, 5H, Ph), 7.56 (d, J = 7.6 Hz, 1H, N–CHCH), 7.73 (s, 1H, NH).
CH), 7.40–7.45 (m, 5H, Ph), 7.56 (d, J = 7.6 Hz, 1H, N–CHCH), 7.73 (s, 1H, NH).
4-[5-(Carboxy)pentylamino]-1-(benzyloxy)pyrimidin-2(1H)-one (3b)..
Into a solution of 2b (590 mg, 1.7 mmol) in 25 mL of MeOH was added 5 mL of 1 M aq. NaOH solution under cooling, and stirred for 10 h. Removal of the solvent and acidification with 1 M HCl to pH 3 resulted in precipitation of 3b (460 mg, 1.4 mmol, 81%): mp 192–193 °C; IR(KBr) 3220–2830, 1710, 1645, 750, 700 cm−1; Anal. Found: C, 61.23; H, 6.52; N, 12.38%. Calcd for C17H21N3O4: C, 61.62; H, 6.39; N, 12.68%; 1H NMR(DMSO-d6) δ 1.31 (br. t, J = 7 Hz, 2H, 3-CH2), 1.44–1.56 (m, 4H, 2,4-CH2), 2.21 (t, J = 7.6 Hz, 2H, CH2CO), 3.18–3.23 (m, 2H, NHCH2), 5.07 (s, 2H, CH2Ph), 5.51 (d, J = 7.6 Hz, 1H, CH), 7.39–7.46 (m, 5H, Ph), 7.56 (d, J = 7.6 Hz, 1H, CHC), 7.70 (br. t, J = 8 Hz, 1H, NH).
4-[5-(Carboxy)pentylamino]-1-(benzyloxy)pyrimidin-2(1H)-one-O-succinimide (4b)..
3b (340 mg, 1.02 mmol) and the water-soluble carbodiimide (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, WSC, 480 mg, 2.5 mmol) was added into 10 mL of a DMF solution of N-hydroxysuccinimide (290 mg, 2.5 mmol) at −10 °C. The solution was kept standing at the same temperature for 1 h and then at room temperature for 14 h. Removal of precipitates and the solvent, followed by addition of water and extraction with CHCl3, eventually gave 4b (420 mg, 0.98 mmol, 96%): mp 186–189 °C; IR(KBr) 1810, 1780, 1740, 1600, 745, 695 cm−1; Anal. Found: C, 6.23; H, 6.02; N, 12.59%. Calcd for C21H24N4O6: C, 61.62; H, 6.39; N, 12.68%; 1H NMR(DMSO-d6) δ 1.38 (br. s, 2H, 3-CH2), 1.51 (br. s, 2H, 2-CH2), 1.64 (br. s, 2H, 4-CH2), 2.67 (br. t, J = 7 Hz, 2H, CH2CO), 2.81 (s, 4H, OCCH2CH2CO), 3.20–3.22 (br. s, 2H, CH2NH), 5.07 (s, 2H, CH2Ph), 5.51 (d, J = 7.6 Hz, 1H, N–CHCH), 7.40–7.45 (m, 5H, Ph), 7.56 (d, J = 7.6 Hz, 1H, N–CHCH), 7.73 (s, 1H, NH). The product was judged pure enough to proceed to the next step.
La-OBn (5a)..
Into 6A,6C,6E-triamino-6A,6C,6E-trideoxy-2A,2B,2C,2D,2E,2F,3A,3B,3C,3D,3E,3F,6B,6D,6F-pentadeca-O-methyl-α-CyD (CyD-NH2,11 420 mg, 0.36 mmol) dissolved in 60 mL of DMF was added 4a (500 mg, 1.3 mmol) and the solution was stirred for 1 day at 40 °C. After removal of the solvent, the residue was subjected to column chromatography on silica gel (CHCl3–MeOH 7∶1 v/v) and 5a was obtained in 28% yield (210 mg, 0.1 mmol): mp 165–168 °C; 1H NMR(DMSO-d6) δ 2.41 (br. s, 6H, CH2CO), 3.00–3.72 (m, 114H, CyD C–H and H2O protons), 4.97 (br. s, 6H, CyD C-1 protons), 5.06 (s, 6H, CH2Ph), 5.58 (d, J = 7.6 Hz, 3H, N–CHCH), 7.39–7.44 (m, 15H, Ph), 7.62 (d, J = 7.2 Hz, 3H, N–CHCH), 7.80 (br. s, 3H, NH); MS(FAB+): m/z 1994 (M + 1) for C93H133N12O36.
Lb-OBn (5b)..
The compound was prepared from 4b and CyD-NH2 according to the same method as mentioned above. The product was obtained in 76% yield (350 mg, 0.16 mmol); mp 147–150 °C; 1H NMR(DMSO-d6) δ 1.29 (br. s, 6H, 3-CH2), 1.47 (br. s, 12H, 2,4-CH2), 2.13 (br. s, 6H, CH2CO), 3.02–3.72 (m, 116H, CyD C-1 protons), 4.96 (br. s, 6H, CyD C-1 protons), 5.06 (s, 6H, CH2Ph), 5.51 (d, 3H, J = 7.2 Hz, 3H, N–CHCH), 7.39–7.43 (m, 15H, Ph), 7.57 (d, J = 7.2 Hz, 3H, N–CHCH), 7.74 (s, 3H, NH); MS(FAB+): m/z 2121 (M + 1) for C102H150N12O36.
Deprotection of the benzyl groups..
10% Pd/C (25 mg) was added into a 20 mL MeOH solution of 5a (200 mg, 0.096 mmol). A stream of hydrogen gas was bubbled into the mixture for 14 h. After disappearance of the starting compound on a TLC plate, Pd/C was filtered and the solvent was distilled off. Dissolving the residue in a small amount of water and lyophilization afforded La in 75% yield: mp 168–170 °C; 1H NMR(DMSO-d6) δ 2.41 (br. s, 6H, CH2CO), 3.05–3.67 (m, 147H, CyD and H2O protons), 4.97 (br. s, 6H, CyD C-1 protons), 5.61 (d, J = 7.6 Hz, 3H, N–CHCH), 7.67–7.82 (m, 6H, NH and N–CHCH); MS(FAB−): m/z 1721 (M − 1) for C72H113N120O36.
Similarly, debenzylation of 5b gave Lb in 75% yield: mp 168–170 °C; 1H NMR(DMSO-d6) δ 1.23 (br. s, 6H, 3-CH2), 1.47 (br. s, 12H, 2,4-CH2), 2.12 (br. s, 6H, CH2CO), 3.00–3.72 (m, 111H, CyD and H2O protons), 4.97 (br. s, 6H, CyD C-1 protons), 5.58 (d, J = 7.6 Hz, 3H, N–CHCH), 7.65 (m, 6H, NH and N–CHCH); MS(FAB−): m/z 1847 (M − 1) for C81H131N12O36. No peaks due to the benzyl protons were observed in either case; the NMR spectra of the debenzylated products were identical with those of the corresponding benzylated derivatives except for the lack of benzyl peaks at 5.06 ppm and at 7.4 ppm. TLC of La and Lb showed a single spot at the starting point, which was colored with Fe3+, indicating that the hydrogenolysis resulted in debenzylation. The products were judged pure enough for titration experiments. Therefore, no further purification was attempted.
Instruments
IR spectra were recorded on a Shimadzu FT-IR 8200 spectrometer. 1H NMR spectra were measured using a Nihondensi Detum α-400 spectrometer operating at 400 MHz. UV–vis and CD spectra were recorded on a Shimadzu UV-2500 PC spectrophotometer and a J-720 circular dichroism spectrophotometer, respectively. The FAB-MS spectra were recorded on a Nihon Bunko JMS-700T spectrometer.
UV–vis and CD titrations
A typical procedure: Titrations were performed in a thermostatted cell holder maintained at 25 °C and the following protocol was typically employed: A stock solution of 54.2 mg La in 1 mL of MeOH, a 5 mL stock solution containing 90.9 mg Fe(NO3)3·9H2O, and a 5 mL stock solution containing 83.9 μL 2,6-lutidine were prepared on the day of the experiment. Stock solutions of 30 μL ligand and 20 μL Fe3+ ([L] = [Fe3+] = 0.3 mM) were added into 3 mL of MeOH in a UV cell. The same samples used for UV measurements were also employed for the CD measurements.
Results and discussion
Complexation of La with Fe3+
A MeOH solution of an equimolar mixture of La and Fe(NO3)3 afforded a UV–vis spectrum involving a maximum absorption band at 490 nm. The absorption at 490 nm demonstrates the existence of di- and/or tetra-coordinate complexes, but not a hexa-coordinate octahedral complex.12Fig. 2 displays the spectra of the MeOH solution in the presence of different concentrations of 2,6-lutidine base. The band maximum shifted with increasing base concentrations eventually getting to 458 nm, which was attributable to the ligand-to-metal charge transfer absorption of the octahedral complex. Thus, to make the reaction go to completion in organic solvents, the addition of a certain amount of base into the system would be required to effectively deprotonate the 1-hydroxy group on the pyrimidinone ring.4 In fact, as one can see from the inset of Fig. 2, at least four equivalents of the base were needed to attain a constant absorbance at 458 nm. Therefore, UV–vis spectra were measured at several different concentrations of Fe3+ in the presence of six equivalents of 2,6-lutidine over the ligand all through this study. The change in absorbance in MeOH with changing molar concentrations of Fe3+ at a constant concentration of La is shown in Fig. 3 and the inset of Fig. 3 displays a titration plot of the absorbance at 458 nm versus Fe3+ concentration which has a sharp break point at a 1∶1 molar ratio, confirming that the existing single species has a stoichiometric composition of 1 equivalent of Fe3+ for 1 equivalent of La expected for a 1∶1 complex.
![Changes in UV–vis absorption spectra for the La–Fe3+ complex in MeOH with altering Fe3+ concentrations. [La] = 0.3 mM, [2,6-lutidine] = 1.8 mM, [Fe3+] = 0–0.41 mM. The inset shows a variation of absorbance at λmax 458 nm as a function of [Fe3+]/[La].](/image/article/2000/P2/a904153b/a904153b-f3.gif) |
| | Fig. 3 Changes in UV–vis absorption spectra for the La–Fe3+ complex in MeOH with altering Fe3+ concentrations. [La] = 0.3 mM, [2,6-lutidine] = 1.8 mM, [Fe3+] = 0–0.41 mM. The inset shows a variation of absorbance at λmax 458 nm as a function of [Fe3+]/[La].
| |
CD spectra were also measured at different concentrations of Fe3+ in the presence of an excess of 2,6-lutidine. Generally speaking, the spectra all show an exciton coupling-type Cotton effect between the pyrimidinone rings, i.e., a positive band at 400 nm and a negative band at 490 nm (called a negative exciton coupling) and an isodichroic point at approximately 440 nm, which is a region where all the traces converge, demonstrates that either one of two possible conformers (a right-hand helicity, Δ and a left-hand helicity, Λ) should be predominant during the titration in these solvents. Evidently, the chiral template α-CyD provides necessary symmetry conditions for distinguishing between right- and left-handed triple helices. CD spectra of the complex in, for example, MeOH and DMSO solvents are comparatively displayed in Fig. 4a and 4b, respectively. The CD band is slightly unsymmetrical due to the cancellation effect of the nearby lying CD band or possibly due to the presence of an unidentified CD band in this region. Furthermore, an observation that the CD intensities are remarkably different in DMSO and MeOH, i.e., much stronger in DMSO than in MeOH, strongly implies that there is a larger difference in configurational energies between two Δ- and Λ-helicates at equilibrium in DMSO vs. MeOH.
In Fig. 5 are collected several plots of CD intensities vs. Fe3+ concentrations in a range of solvents. The fact that the La complex shows a negative exciton coupling indicates that the complex adopts predominantly a Δ-helix in all the solvents examined. The amplitudes of the signals at a selected wavelength are summarized in Table 1 together with UV–vis data. The absolute magnitude of the signals with aprotic solvents increased in the order: THF ≤ MeCN < Me2CO < HCONH2 < DMF < DMSO, indicative of the importance of their hydrogen-bond accepting properties (β-values) rather than their overall solvent polarity such as ET and (ε − 1)/(2ε + 1) values.13 This observation strongly suggests that both amido NH and exocyclic NH groups in the La–Fe3+ complex should protrude from the cavity so as to be capable of hydrogen-bonding to the bulk solvents. Particularly, the exocyclic NH groups, as consideration of space-filling CPK molecular models suggests, are unable to enter into the cavity owing to steric crowding in its inner space. In order to give insight into the mechanism of the observed solvent effects on the expression of the helicity, Fig. 6 displays schematic illustrations of the parts of the La–Fe3+ complexes in the Δ- and Λ-states; the spacer methylenes and the linker amido group are less restricted to rotational freedom for the Δ-triple stranded helicate than for the Λ-helicate; namely, the Δ-helicate has an extended, less sterically congested, and thereby entropically favorable structure, supporting the proposal that the 1-pyrimidinone complex with a negative exciton coupling has Δ-configuration, as does the naturally occurring siderophore enterobactin. Incidentally, CD signals observed in the case of hydroxylic solvents including water are relatively weak in spite of their strong hydrogen-bond accepting ability, where the same helical propensity is still retained. This unequivocally implies that the strong hydrogen-bond accepting abilities of the hydroxylic solvents appear to be almost cancelled by their hydrogen-bond donating abilities.
Table 1 UV–vis and CD data for the La– and Lb–Fe3+ complexes in various solvents
| |
La
|
Lb
|
| Solvent |
λ
max (Abs)/nm |
CD intensitya/mdeg |
λ
max (Abs)/nm |
CD intensitya/mdeg |
|
At 480 nm.
|
| DMSO |
462.0 (1.51) |
−7.65 |
469.0 (1.59) |
13.1 |
| DMF |
462.0 (1.53) |
−5.0 |
467.0 (1.60) |
10.0 |
| HCONH2 |
463.5 (1.39) |
−4.5 |
469.5 (1.45) |
7.1 |
| THF |
461.0 (1.35) |
−0.45 |
467.0 (1.40) |
4.6 |
| MeCN |
458.5 (1.41) |
−0.72 |
458.5 (1.44) |
3.0 |
| Me2CO |
459.5 (1.44) |
−1.25 |
459.5 (1.50) |
1.0 |
| H2O |
456.0 (1.38) |
−0.41 |
462.5 (1.49) |
0.0 |
| BuOH |
460.0 (1.39) |
−0.78 |
463.5 (1.45) |
1.8 |
| EtOH |
458.5 (1.42) |
−3.10 |
463.0 (1.46) |
−1.8 |
| MeOH |
458.0 (1.39) |
−3.80 |
460.0 (1.43) |
−2.5 |
![Variations of CD intensities at 480 nm as a function of [Fe3+]/[La] in various solvents. [La] = 0.45 mM, [2,6-lutidine] = 2.7 mM, [Fe3+] = 0–0.62 mM.](/image/article/2000/P2/a904153b/a904153b-f5.gif) |
| | Fig. 5 Variations of CD intensities at 480 nm as a function of [Fe3+]/[La] in various solvents. [La] = 0.45 mM, [2,6-lutidine] = 2.7 mM, [Fe3+] = 0–0.62 mM.
| |
 |
| | Fig. 6 Schematic representation of favorable induction of a Δ- over Λ-helicity around Fe3+ in the La–Fe3+ complex. For simplicity only one of the three side chains is shown. The delta (δ) denotes presumable charge separation.
| |
Complexation of Lb with Fe3+
As Fig. 7 shows, the ligand Lb exhibited the same stoichiometric behavior as did La to form a Lb–Fe3+ 1∶1 complex, which was 25-fold less stable toward removal of Fe3+ by EDTA than the La complex due probably to an entropic reason (Fig. 8).
![Variation of UV–vis absorbance at 460 nm as a function of [Fe3+]/[Lb] in MeOH. [Lb] = 0.3 mM, [2,6-lutidine] = 1.8 mM, [Fe3+] = 0–0.41 mM.](/image/article/2000/P2/a904153b/a904153b-f7.gif) |
| | Fig. 7 Variation of UV–vis absorbance at 460 nm as a function of [Fe3+]/[Lb] in MeOH. [Lb] = 0.3 mM, [2,6-lutidine] = 1.8 mM, [Fe3+] = 0–0.41 mM.
| |
![Comparison of competitive Fe3+-removal by EDTA from the La–, Lb–, and DFB–Fe3+ complexes in MeOH. [La] = [Lb] = [DFB] = [Fe3+] = 0.3 mM, [2,6-lutidine] = 2.7 mM, [EDTA] = 0–9.6 mM. DFB: desferrioxamine B.](/image/article/2000/P2/a904153b/a904153b-f8.gif) |
| | Fig. 8 Comparison of competitive Fe3+-removal by EDTA from the La–, Lb–, and DFB–Fe3+ complexes in MeOH. [La] = [Lb] = [DFB] = [Fe3+] = 0.3 mM, [2,6-lutidine] = 2.7 mM, [EDTA] = 0–9.6 mM. DFB: desferrioxamine B.
| |
The CD spectra of the Lb complex are of particular interest for comparison of its performance with the La complex. The CD data are summarized in Fig. 10 and Table 1. The Lb complex in highly polar aprotic solvents such as DMF and DMSO exhibited an unexpectedly strong positive exciton coupling with a crossover at 435 nm (Fig. 9b), in spite of its bearing long pentamethylene spacers with high conformational flexibility at the periphery of the coordination site. The positive exciton coupling implies a complexation with a Λ-helix with the same handedness as that for the naturally occurring ferrichrome. On the other hand, MeOH and EtOH provided only a weak negative exciton coupling (Fig. 9a), and water no exciton coupling. In Fig. 11 are given plots of CD intensities versus the hydrogen-bond accepting properties (β-values) of the solvent.13 The approximate linearity within aprotic solvents suggests that the induced helicity primarily arises from hydrogen-bonding between the solvent and the amido N–H group rather than the solvent and the amide CO group (Fig. 11).
![Variations of CD intensities at 480 nm as a function of [Fe3+]/[Lb] in various solvents. [Lb] = 0.45 mM, [2,6-lutidine] = 2.7 mM, [Fe3+] = 0–0.62 mM.](/image/article/2000/P2/a904153b/a904153b-f10.gif) |
| | Fig. 10 Variations of CD intensities at 480 nm as a function of [Fe3+]/[Lb] in various solvents. [Lb] = 0.45 mM, [2,6-lutidine] = 2.7 mM, [Fe3+] = 0–0.62 mM.
| |
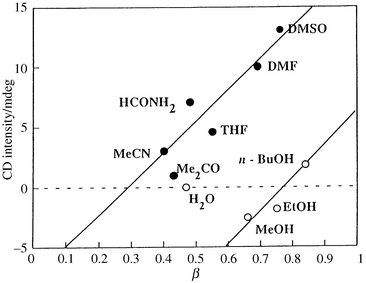 |
| | Fig. 11 Plots of CD intensities of the Lb complex at 480 nm versusβ values, which are the hydrogen-bond accepting abilities of the solvents. The line is arbitrarily drawn within aprotic solvents (open circles).
| |
Furthermore, it should be noted that the amplitudes of the CD spectra of the Lb complex in aprotic solvents were even larger than those of the corresponding La complex, although, as CPK models show, all the segments in the side-chains can rotate freely with no inherent barriers; such rotations seem not advantageous for any chiral induction.
Careful inspection of the CPK models reveals that a cooperative combination of two different hydrogen-bonds would prevail in constructing an overall structure of the side chain leading to a large CD amplitude; as illustrated in Fig. 12, the linker amido N–H group is forced to protrude from the inner cavity by a hydrogen-bonding interaction with a bulk solvent molecule (a), while the linker amido carbonyl group is oriented to the inner space of the cavity to form a hydrogen-bond with the exocyclic amino N–H group within the same chain (b). The hydrogen-bond between the exocyclic N–H and amido CO groups is more preferred in the Λ-helicate than in the Δ-helicate, because the H⋯O–C hydrogen-bond angle of 120–150° in the Λ-helix is more favorable than that of less than 90° in the Δ-helix. Conveniently, the hydrogen-bonds thus formed would be largely stabilized by π-electron resonance between the exocyclic amino-nitrogen lone-pair and the electron-withdrawing pyrimidinone group (Scheme 2), and thereby would direct the side-chain configuration towards a rigid propeller-like arrangement; namely, the enhanced helical propensity in aprotic solvents appears to be attributable in part to the stabilized intramolecular hydrogen-bond and in part to lack of a solvent molecule competing for the exocyclic amino NH groups located in the hydrophobic inner space. Shanzer and his co-workers suggested the importance of formation of extended hydrogen-bond networks in an iron complex; these hydrogen-bonds restrict the conformations to affect the relative stability of the diastereomers and to stabilize the complex once formed.14 However, such a helical structure would be easily disrupted when the intramolecular hydrogen-bonds are cleaved by a hydroxylic solvent. Actually, no exciton coupling was observed in water. Usually, exciton couplings in alcohols are only small, and negative in MeOH and EtOH but positive in n-BuOH, demonstrating that the configuration preferentially adopted by the Lb–Fe3+ complex depends on the alcoholic solvents employed. Thus, the screw sense of the Lb complex in alcohols is somewhat difficult to implicate owing to the complexing nature of hydroxylic solvents. Further study would be needed to explain the cause of the configurational preference for a Δ-helix in MeOH, and for a Λ-helix in n-BuOH.
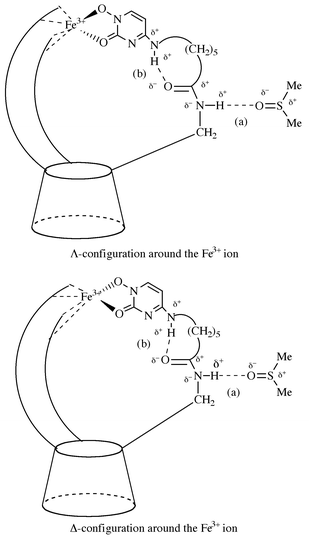 |
| | Fig. 12 Schematic representation of favorable induction of a Λ- over Δ-helicity around Fe3+ in the Lb–Fe3+ complex. For simplicity only one of the three side chains is shown. The delta (δ) denotes presumable charge separation.
| |
 |
| | Scheme 2 | |
Conclusions
α-CyD-based, C3-symmetric, and tripodal ferrichrome mimics with different spacers were prepared and their spectroscopic behavior was examined to investigate the causes of induced helicities. La having short spacers exhibited a negative exciton coupling, showing a Δ-helicity in all solvents tested, while Lb having long spacers formed a Λ-helicate with an unexpectedly high chirality in highly aprotic solvents such as DMSO and DMF. It became evident that the flexibility of the side chains determines the helicity of the former complex, while intramolecular hydrogen-bonding is substantial for helical induction in the latter complex.
References
-
K. N. Raymond
and J. R. Telford
, Siderophore-Mediated Iron Transport in Microbes, in Bioinorganic Chemistry, ed. D. P. Kessissoglou, Dordrecht Kluwer, 1995, p. 225;
Search PubMed; K. N. Raymond, G. Mueller and B. F. Matzanke, Top. Curr. Chem., 1984, 123, 49 Search PubMed.
-
J. B. Neilands
, T. Peterson
and S. A. Leong
, High Affinity Iron Transport in Microorganisms, in Inorganic Chemistry in Biology and Medicines, ed. A. E. Martell, American Chemical Society, Washington, DC, 1980, p. 263;
Search PubMed;
C. G. Pitt
and A. E. Martell
, The Design of Chelating Agents for the Treatment of Iron Overload, ibid., p. 279;
Search PubMed;
Y. Sun
and A. E. Martell
, The Development of Iron Chelators for Clinical Use, ed. R. J. Bergeron and G. M. Brittenham, CRC Press, Boca Raton, 1992, p. 345.
Search PubMed.
- Y. Tor, J. Libman and A. Shanzer, J. Am. Chem. Soc., 1987, 109, 6518 CrossRef CAS; A. Shanzer, J. Libman, R. Lazar, Y. Tor and T. Emery, Biochem. Biophys. Res. Commun., 1988, 157, 389 CAS; E. Jurkevitch, Y. Hadar, Y. Chen, J. Libman and A. Shanzer, J. Bacteriol., 1992, 174, 78 CAS; M. Akiyama, A. Katoh, J. Kato, K. Takahashi and K. Hattori, Chem. Lett., 1991, 1189 CAS.
- R. Nudelman, O. Ardon, Y. Hadar, Y. Chen, J. Libman and A. Shanzer, J. Med. Chem., 1998, 41, 1671 CrossRef CAS; J. Ohkanda and A. Katoh, Rev. Heteroatom Chem., 1998, 18, 87 Search PubMed.
- Y. Hori, J. Hayashi and S. Tamagaki, J. Chem. Soc., Chem. Ind. Chem., 1998, 417 Search PubMed.
- Y. Hori and S. Tamagaki, J. Chem. Soc., Chem. Ind. Chem., 1998, 602 Search PubMed.
- J. Ohkanda, T. Tokumitsu, K. Mitsuhashi and A. Katoh, Bull. Chem. Soc. Jpn., 1993, 66, 841 CAS; Y. Hida, T. Konakahara and A. Katoh, J. Org. Chem., 1997, 62, 3618 CrossRef.
- K. P. Meurer and F. Voegtle, Top. Curr. Chem., 1985, 1 Search PubMed; I. W. Zarges, J. Hall, J.-M. Lehn and C. Bolm, Helv. Chim. Acta, 1991, 74, 1843 CrossRef; J. H. Brewster, Top. Curr. Chem., 1974, 47, 29 Search PubMed; C. R. Woods, M. Benaglia, F. Cozzi and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1830 CrossRef CAS.
- J. Ohkanda, J. Kamitani, T. Tokumitsu, Y. Hida, T. Konakahara and A. Katoh, J. Org. Chem., 1997, 62, 3618 CrossRef.
- M. R. Harden, L. J. Jennings and A. Parkin, J. Chem. Soc., Perkin Trans. 1, 1990, 2175 RSC; W. Klotzer, Monatsh. Chem., 1964, 95, 1729.
- S. L. Legen, S. Quici and M. D. Ryan, J. Am. Chem. Soc., 1979, 101, 7630 CrossRef.
- S. Blanc, P. Yakirevitch, E. Leize, M. Meyer, J. Libman, A. V. Dorsselaer, A. M. Albrecht and A. Shanzer, J. Am. Chem. Soc., 1997, 119, 4934 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 410 RSC.
- J. Libman and Y. Tor, J. Am. Chem. Soc., 1987, 109, 5880 CrossRef CAS; A. Shanzer, J. Libman and S. Lifson, Pure Appl. Chem., 1992, 64, 1421 CAS; I. Dayan, J. Libman, Y. Agi and A. Shanzer, Inorg. Chem., 1993, 32, 1467 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here. ![Changes in UV–vis absorption spectra for the La–Fe3+ complex in MeOH with various concentrations of 2,6-lutidine. [La] = [Fe3+] = 0.3 mM, [2,6-lutidine] = 0 –1.56 mM. The inset shows a plot of λmaxversus [2,6-lutidine]/[La].](/image/article/2000/P2/a904153b/a904153b-f2.gif)
![CD titration of La with Fe3+ in a) MeOH and b) DMSO. [La] = 0.45 mM, [2,6-lutidine] = 2.7 mM, [Fe3+] = 0–0.62 mM.](/image/article/2000/P2/a904153b/a904153b-f4.gif)
![CD titration of Lb with Fe3+ in a) MeOH and b) DMSO. [Lb] = 0.45 mM, [2,6-lutidine] = 2.7 mM, [Fe3+] = 0–0.62 mM.](/image/article/2000/P2/a904153b/a904153b-f9.gif)