Three approaches to the synthesis of L-leucine selectively labelled with carbon-13 or deuterium in either diastereotopic methyl group
Matthew D.
Fletcher
a,
John R.
Harding
b,
Rachael A.
Hughes
a,
Nicholas M.
Kelly
a,
Holger
Schmalz
a,
Andrew
Sutherland
a and
Christine L.
Willis
a
aSchool of Chemistry, University of Bristol, Cantock’s Close, Bristol, UK BS8 1TS
bZeneca Pharmaceuticals, Mereside, Alderley Park, Macclesfield, UK SK10 4TG
First published on 12th January 2000
Abstract
Three approaches to the synthesis of L-leucine selectively labelled with carbon-13 or deuterium in either diastereotopic methyl group as well as at C-3 and C-4 are described. In all three methods the stereogenic centre at C-2 was created with total stereocontrol via a one-pot, two-enzyme catalysed procedure involving hydrolysis and reductive amination of a 2-keto ester. However, the approaches vary in the synthesis of the isotopically labelled 2-keto esters and in the production of the stereogenic centre at C-4 which was achieved either via alkylation of a propionylated Evans’ auxiliary with labelled iodomethane or by the diastereoselective conjugate addition of a labelled organocopper reagent to crotonate tethered to a chiral sultam. The latter approach proved most efficient and using the (1R,2S,3R)-3-[N-phenylsulfonyl-N-(3,5-dimethyldiphenyl)aminobornan-2-ol ester 27, [5-13C]-L-leucine was prepared with >98% de at C-4 and in 49% overall yield from the first labelled intermediate 28.
Introduction
Isotopically labelled L-amino acids serve a vital role in a variety of studies in bioorganic chemistry.1,2 For example, the selective labelling of either diastereotopic methyl group of L-leucine has enabled the elucidation of their fate during secondary metabolic reactions.3–7 Leucine is also important in protein tertiary structure formation due to its participation in hydrophobic interactions. Assignment of the leucine pro-R and pro-S methyl group resonances in protein NMR spectra allows more precise definition of protein structure and the study of stereochemical aspects of associated molecular recognition phenomena.8,9 Thus several groups have investigated approaches to the synthesis of leucine selectively labelled in either diastereotopic methyl group for example via biosynthesis,10,11 using chemical and enzymatic resolutions,5,6,7,11,12 and using starting materials from the chiral pool.13 Young and co-workers performed the first fully stereoselective synthesis of (2S,4R)-[5,5,5-2H3]-leucine using (2S)-pyroglutamic acid as a chiral template![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 14 and more recently a similar approach has been reported by Nishiyama and co-workers.15 We have previously described a chemo-enzymatic route for the synthesis of L-leucine selectively labelled with deuterium and/or carbon-13 in either diastereotopic methyl group.16 Our strategy was versatile enabling the incorporation of isotopic labels at C-3 and C-4 as well as in either diastereotopic methyl group and gave total stereocontrol at C-2. However, there were technical difficulties in handling the volatile intermediates and only 86% de at C-4 was achieved. We now describe this approach in full as well as our more recent investigations which have led to the development of a new and efficient strategy for the synthesis of isotopically labelled L-leucine which not only overcomes the problems of handling volatile intermediates but also gives >98% de at C-4.
14 and more recently a similar approach has been reported by Nishiyama and co-workers.15 We have previously described a chemo-enzymatic route for the synthesis of L-leucine selectively labelled with deuterium and/or carbon-13 in either diastereotopic methyl group.16 Our strategy was versatile enabling the incorporation of isotopic labels at C-3 and C-4 as well as in either diastereotopic methyl group and gave total stereocontrol at C-2. However, there were technical difficulties in handling the volatile intermediates and only 86% de at C-4 was achieved. We now describe this approach in full as well as our more recent investigations which have led to the development of a new and efficient strategy for the synthesis of isotopically labelled L-leucine which not only overcomes the problems of handling volatile intermediates but also gives >98% de at C-4.
Results and discussion
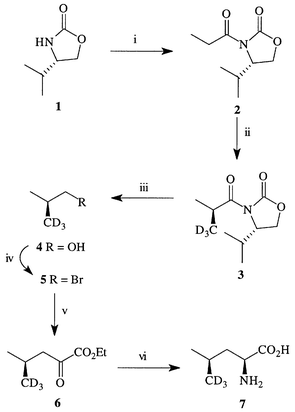
Method 1. Synthesis of (2S,4R)-[5,5,5-2H3]leucine 7
Our initial route16 to the synthesis of L-leucine selectively labelled with deuterium and/or carbon-13 in either diastereotopic methyl group is shown in Scheme 1. First it was necessary to create the stereogenic centre which was to become C-4 in L-leucine and this was formed via alkylation of the known17,18 propionylated oxazolidinone 2 with CD3I to give 3 (or 13CH3I to give 8, Scheme 2). The optimum conditions for the reaction proved to be generation of the enolate with sodium hexamethyldisilazide (NaHMDS) and quenching with 10 equivalents of iodomethane giving the product in 90% yield. However, to minimise the quantity of expensive isotopically labelled iodomethane, further optimisation showed that 1.4 equivalents of 13CH3I giving 76% yield of 8 was the best compromise. The alternative diastereomer 11 was prepared from (4S)-3-acetyl-4-isopropyloxazolidinone 9 by an initial alkylation with 13CH3I followed by alkylation with iodomethane (Scheme 2). In the 1H NMR spectra of 8 and 11 the signals assigned to 2′-CH3 and 2′-13CH3 were well resolved due to the large 13C–1H coupling and integration of the signals at δ 1.22 (dd, J 128.3, 6.9 Hz in 8; dd, J 6.9, 5.0 Hz in 11) and at δ 1.15 (dd, J 6.7, 5.2 Hz in 8; dd, J 127.9, 6.7 Hz in 11) confirmed that selective alkylation of (4S)-3-propionyloxazolidinone had occurred giving a 13∶1 mixture of diastereomers.
 | ||
| Scheme 1 Reagents: i, BuLi, EtCOCl, 94%; ii, NaHMDS, CD3I, 72%; iii, LiAlH4, Et2O; iv, Ph3P, Br2, PhNO2; v, Mg, (CO2Et)2, Et2O, THF, −78 °C, 14–60% from 3; vii, CRL, leucine dehydrogenase, FDH, HCO2NH4, 85%. | ||
 | ||
| Scheme 2 | ||
With the required stereogenic centre established, the next stage was to cleave the auxiliary and to effect a two-carbon homologation to the 2-keto ester 6. Many methods are known for the synthesis of 2-keto esters and we favoured the reaction of a Grignard reagent with diethyl oxalate at low temperature.19 Reduction of 3 with lithium aluminium hydride cleaved the chiral auxiliary and liberated the labelled alcohol 4 which was converted to the bromide 5 using triphenylphosphine and bromine. Both alcohol 4 (bp20ca. 108 °C) and bromide 5 (bp20ca. 91 °C) are rather volatile necessitating the use of manifold trap techniques for their efficient manipulation. Nitrobenzene was used as the solvent in the conversion of 4 to 5 so that bromide 5 could be distilled from the reaction mixture and then used immediately to form the Grignard reagent, which was reacted in situ with diethyl oxalate to give the 2-keto ester 6 in variable yields (14–60% from 3).
The final stages of the synthesis involved hydrolysis of the ester and the reductive amination of the resultant 2-keto acid catalysed by a commercially available amino acid dehydrogenase. These enzymes require NADH; the cofactor may be recycled efficiently in situ according to the protocol of Shaked and Whitesides using a second commercially available enzyme, formate dehydrogenase (FDH), with the consumption of formate ions and the evolution of carbon dioxide.21 In our initial work16 we converted α-keto ester 6 to the acid by saponification with sodium hydroxide. More recently we have adopted a milder procedure using a lipase isolated from Candida rugosa (CRL), as model reactions showed that it provides higher yields than saponification. Furthermore the reaction conditions of the lipase hydrolysis are compatible with those of the leucine dehydrogenase catalysed reductive amination.22 Thus a one-pot dual enzyme catalysed hydrolysis of 2-keto ester 6 and reductive amination of the resultant 2-keto acid gave (2S,4R)-[5,5,5-2H3]-leucine 7 in 85% yield from 6. The methyl region of the 1H NMR spectrum of 7 displayed only the doublet corresponding to the lower frequency methyl resonance of unlabelled L-leucine; this is consistent with previous assignments of the lower frequency resonance to the 4-pro-S and the higher frequency resonance to the 4-pro-R leucine methyl groups.12,14 The 13C NMR spectrum of (2S,4R)-[5,5,5-2H3]leucine 7 showed a singlet at δC 20.8 due to the methyl group and a quintet at δC 20.7, assigned to the trideuteromethyl group. On first inspection these are confusing observations as, in common with the 1H NMR spectrum, the lower frequency 13C resonance has been assigned previously to the 4-pro-S and the higher frequency resonance to the 4-pro-R leucine methyl groups;10,12,14 however, the apparent discrepancy is caused by an α-isotope shift23 of the trideuteromethyl group resonance, as noted by Young and co-workers.14
In principle this approach may be used for the synthesis of the complementary diastereomer, (2S,4S)-[5,5,5-2H3]leucine by the route shown in part in Scheme 2 (using CD3I). A more direct route would use the (+)-norephedrine derived auxiliary;17 however we found that in this case the key alkylation step proceeds in lower yield than with the L-valine derived auxiliary 1. Therefore for the synthesis of (2S,4S)-[5,5,5-2H3]leucine we suggest the use of the D-valine derived auxiliary (i.e. the enantiomer of 1). This chemo-enzymatic approach to the synthesis of L-leucine has the further advantage that it enables the incorporation of carbon-13 at C-3 and/or C-4 using sodium [13C]acetate as the source of isotopic label (Scheme 2). For example, treatment of sodium [2-13C]acetate with pivaloyl chloride gives a mixed anhydride which on reaction with the lithium salt of 1 gives the acylated product 12 in 70% yield, the precursor to [4-13C]-L-leucine. In addition, the approach may also be modified for the inclusion of nitrogen-15.24
Method 2. Synthesis of (2S,4R)-[5-13C]leucine 17
When designing routes to the synthesis of expensive isotopically labelled compounds it is essential that each stage is high yielding and reproducible. Although the above approach is versatile and most of the steps are indeed high yielding and simple to perform, the route is not ideal due to the problems inherent in the preparation and purification of the volatile bromide 5. Hence we required an alternative strategy for the two-carbon homologation of 2-methylpropanol to the key 2-keto ester. Our aim was to convert the alcohol to a good leaving group such as a sulfonate ester for coupling with the Umpolung 2-keto ester equivalent 2-ethoxycarbonyl-1,3-dithiane25 and finally hydrolysis of the resultant dithiane.
First the carbon-13 labelled derivative 8 was converted into alcohol 13 using a two-step lithium hydroperoxide cleavage26
– reduction sequence (Scheme 3), which gave a better yield than the direct reduction of 8 with lithium aluminium hydride and easy recovery of the Evans’ auxiliary 1 by acid–base extraction after the first step. Model reactions were carried out in order to determine the best mode of activation for the hydroxy group of alcohol 13 for reaction with the salt of 2-ethoxycarbonyl-1,3-dithiane. We found that the toluene-p-sulfonate, methanesulfonate and p-bromobenzenesulfonate all proved insufficiently reactive whereas the bromide and triflate were suitable leaving groups. Given the difficulties we experienced when using bromide 5 in Method 1, we chose triflate 14 as the alkylating agent. To our knowledge, the only previous example of the alkylation of a glyoxylic acid dithioacetal derivative with a triflate is found in Shiba and co-workers’ synthesis of 3-deoxy-D-manno-2-octulosonic acid (KDO).27 Treatment of alcohol 13 with triflic anhydride, pyridine and carbon tetrachloride at 0 °C gave the required triflate 14 in 73% yield. The triflate 14 was used to alkylate 2-ethoxycarbonyl-1,3-dithiane (CBT) to provide 2-ethoxycarbonyl-2-{(2R)-[3-13C]-2-methylpropyl}-1,3-dithiane 15. It was not necessary to fully purify 15 and oxidative hydrolysis of crude 15 with N-bromosuccinimide (NBS)25,28 gave, after column chromatography, pure 2-keto ester 16 in 45% yield over the two steps.
 | ||
| Scheme 3 Reagents: i, a, LiOH, H2O2; b, LiAlH4, Et2O, 88%; ii, Tf2O, pyridine, CCl4, 73%; iii, BuLi, CDT, 56%; iv, NBS, Me2CO, H2O, 80%; v, CRL, leucine dehydrogenase, FDH, HCO2NH4, 79%. | ||
2-Keto ester 16 was converted directly into (2S,4R)-[5-13C]leucine 17 in 79% yield, using our one-pot two-step hydrolysis–reductive amination sequence. As expected, the methyl region of the 1H NMR spectrum of (2S,4R)-[5-13C]leucine 17 featured two signals with the downfield signal (δ 0.95) displaying the larger coupling (J 125 Hz) to carbon-13. The 13C NMR spectrum displayed one strongly enriched signal (δ 22.8) and, at higher field (δ 21.7), a weakly enriched signal {due to the minor diastereomer, (2S,4S)-[5-13C]leucine 25}, consistent with previous assignments of the leucine methyl groups.10,12,14
Method 3. (2S,4S)-[5-13C]leucine 25
Although the second route to the synthesis of isotopically labelled L-leucine does overcome the problem of handling 1-bromo-2-methylpropane and each step of the synthesis is reliable and simple to perform, it is still not ideal. The diastereoselectivity at C-4 is only 86% and the overall yield for the synthesis of 2-keto ester 16 from the first isotopically labelled intermediate 8 is approximately 30%. Hence our third and final approach aimed to address these deficiencies and used different disconnections from the preceding two methods: the pivotal 2-keto ester came from a one-carbon homologation of labelled 3-methylbutanoic acid (isovaleric acid), itself ultimately derived from crotonic acid (Scheme 4). | ||
| Scheme 4 | ||
We proposed to synthesise the stereoselectively labelled 3-methylbutanoic acid by conjugate addition of a labelled organometallic reagent to a crotonic acid derivative, with a chiral auxiliary inducing the stereoselectivity. The application of Evans’ type auxiliaries in this context is uncommon,18,29,30 although some success has been reported both by Hruby and co-workers31 and Williams and co-workers.32 In contrast, Oppolzer and co-workers’ camphorsultam 18 is well documented as a suitable chiral auxiliary for conjugate additions.33 Method 3 therefore began with the crotonylation of (1S)-1,10-camphorsultam 1833b to provide the conjugate addition substrate 19 in 84% yield (Scheme 5).
 | ||
| Scheme 5 Reagents: i, NaH, crotonyl chloride, 84%, ii, (13CH3)2CuLi·PBu3, 89%; iii, H2O2, LiOH, 93%; iv, Ph3PCHCN 22, EDCI, DMAP 80%; v, O3, MeOH, CH2Cl2, 76%; vi, CRL, leucine dehydrogenase, FDH, HCO2NH4, 81%. | ||
Oppolzer and co-workers have reported the highly diastereoselective conjugate addition of Grignard reagents and organocuprates to substrates similar to 19.33 However the addition of a methyl group is something of a special case: problems of 1,2 addition are suffered with Grignard reagents33c and low π-face discrimination with copper(I) catalysed Grignard reagents.33e Oppolzer and co-workers minimised these difficulties by employing phosphine stabilised Gilman reagents (3 equivalents), which give good regio- and stereo-selectivity.33d,e We needed to further optimise the conditions of our conjugate addition for we required not only maximum yield and maximum diastereoselectivity but also maximum isotope yield. Our investigations revealed that reduction of the quantity of Gilman reagent below 2.6 equivalents significantly lowered the reaction yield, as might be expected from inspection of the proposed mechanism.33d Our optimised conditions were thus treatment of chiral crotonyl derivative 19 with 2.6 equivalents of labelled Gilman reagent {prepared from [13C]iodomethane, lithium metal and copper(I) iodide–tri-n-butylphosphine complex} at −78 °C, which gave the conjugate addition product 20 in 89% yield. The diastereoselectivity of this reaction was determined from integration of the signals assigned to 3′-CH3 (δ 0.96, dd, J 6.6, 5.5 Hz) and 3′-13CH3 (δ 0.97, dd, J 125.2, 6.6 Hz) in the 1H NMR spectrum which indicated that a 10∶1 mixture of diastereomers (ca. 82% de) had been formed.
The auxiliary was cleaved with lithium hydroperoxide33g (and recovered in 85% yield) to liberate (S)-[4-13C]-3-methylbutanoic acid 21 in 93% yield. The one-carbon homologation of 21 was achieved via formation and ozonolysis of the β-ketocyanophosphorane 23 under the conditions described by Wasserman and Ho34 giving methyl (S)-[5-13C]-4-methyl-3-oxopentanoate 24. Finally the one-pot two-step enzymatic catalysed conversion of 2-keto ester 24 to (2S,4S)-[5-13C]leucine 25 proceeded smoothly giving an 81% yield of pure product after ion exchange chromatography. This approach may be readily adapted to the synthesis of the complementary diastereomer (2S,4R)-[5-13C]leucine 17 by starting from the commercially available (1R)-1,10-camphorsultam. This approach to the synthesis of either (2S,4S)- or (2S,4R)-[5-13C]leucine is high yielding and each step is simple to perform and reliable. The only downside of this route was the poor diastereoselectivity at C-4; thus we sought to improve this by using a different chiral auxiliary.
Helmchen and co-workers have described the use of (1R,2S,3R)-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 26 as a (commercially available) chiral auxiliary for conjugate addition reactions.35 Acylation of chiral auxiliary 26 with crotonyl chloride35b gave enoate 27 in 81% yield (Scheme 6). As in the case of the Oppolzer auxiliary, we wished to optimise the conjugate addition conditions to produce maximum de and minimum isotopic label wastage. Following Helmchen and Wegner we employed the Yamamoto reagent,36,3713CH3Cu·BF3, in 7-fold excess (a 5-fold excess dramatically reduced the yield), derived from the organolithium with ether as solvent.35a Our optimum conditions gave the conjugate addition product 28 in 79% yields; inclusion of tri-n-butylphosphine (to dissolve and stabilise the Yamamoto reagent)36,38 gave a similar result. NMR spectroscopy (in deuterochloroform, deuteroacetone or deuterobenzene) failed to detect the minor diastereomer (NMR spectroscopy of the conjugate addition product formed under sub-optimum reaction conditions revealed that the signals from each diastereomer’s 3′-CH3 and 3′-13CH3 are partially resolved) so we have achieved our objective and increased the de of the conjugate addition to above 98%.
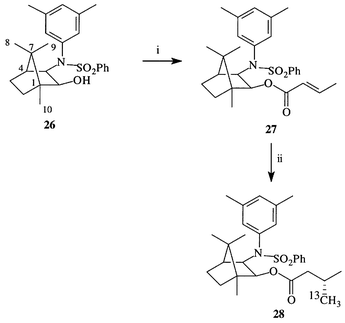 | ||
| Scheme 6 Reagents: i, Crotonyl chloride, molecular sieves, 81%; ii, 13CH3Cu·BF3, 79%. | ||
The auxiliary was cleaved with aqueous potassium hydroxide in methanol35a (and recovered in 93% yield) to liberate (R)-[4-13C]-3-methylbutanoic acid in quantitative yield. Thus, using Helmchen’s auxiliary, we have improved Method 3 to produce a synthesis of (2S,4R)-[5-13C]leucine 17 with greater than 98% de and 39% overall yield from the first labelled intermediate 28. The complimentary diastereomer may be synthesised starting from the commercially available auxiliary (1R,2R,3S)-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]-2-bornanol.35a
Conclusions
We have described three enantioselective syntheses of L-leucine that permit the selective labelling of either diastereotopic methyl group with carbon-13 or deuterium. These routes are extremely versatile and allow incorporation of isotopic labels at other sites and the appropriate method may be chosen for maximum de or maximum isotope yield. We consider the conjugate addition route (Method 3 using Helmchen’s auxiliary) the best of our three methods due to its comparative simplicity, high yields and excellent de.Experimental
General experimental details have been described previously.39 Cuprate reactions were conducted under argon. Copper(I) iodide was purified according to Taylor and Casy.40 Copper(I) iodide–tri-n-butylphosphine complex was prepared according to the procedures of Taylor and Casy40 and Kauffman and Teter41 from purified copper(I) iodide and freshly distilled tri-n-butylphosphine in 66% yield; mp 74–75 °C (lit.,41 75 °C). All NMR spectra were run in CDCl3 unless otherwise stated. J values are in Hz. [α]D has units of 10−1 deg cm2 g−1. The enzymes were purchased and stored as follows: lipase from Candida rugosa (CRL), Sigma, stored at 4 °C as a 10000 eU ml−1 solution in tris buffer (5 mM); formate dehydrogenase (FDH) from Candida boidinii, Boerhinger, stored at 4 °C; leucine dehydrogenase from Bacillus species, Sigma, stored at −20 °C; β-nicotinamide adenine dinucleotide hydride (NADH), Genzyme, stored at −20 °C.
General procedure for the one-pot hydrolysis and reductive amination of 2-keto esters
Potassium phosphate buffer (5 mM, 25 mL per mmol of substrate) was deoxygenated by bubbling through nitrogen for 0.5 h. CRL (10000 eU per mmol of substrate) and the keto ester (1 eq.) in ethanol (1 mL per mmol of substrate) were added and the reaction mixture stirred at rt, maintaining the pH between 7.0 and 8.5 by the periodic addition of sodium hydroxide (1.0 or 0.1 M). Once the pH had stopped changing (ca. 5 h, ca. 1 eq. of sodium hydroxide having been added), ammonium formate (10 eq.), 1 M dithiothreitol (DTT) (1 μL per mmol of substrate), formate dehydrogenase (8 mg, 4 eU for less or 16 mg, 8 eU for more than 3 mmol of substrate), NADH (8 mg, 12 μmol for less or 16 mg, 24 μmol for more than 3 mmol of substrate) and leucine dehydrogenase (5 eU for less or 10 eU for more than 3 mmol of substrate) were added. The resultant solution was stirred at rt and maintained between pH 7.0 and 7.5 by the periodic addition of hydrochloric acid (1.0 or 0.1 M) until the pH remained static (ca. 7 days, ca. 0.8 eq. of hydrochloric acid having been added). The reaction mixture was concentrated in vacuo and the product isolated by ion exchange chromatography on Dowex® 50WX8-100 (20 g per mmol of substrate), eluting first with water (3 × 40 mL per mmol of substrate) then conc. ammonia (2 × 100 mL per mmol of substrate). The conc. ammonia eluent was evaporated to dryness to yield the pure α-amino acid.
The above reaction was repeated using [13C]iodomethane as the electrophile to give a 13∶1 mixture of (4S)-4-isopropyl-3-{(2R)-[3-13C]-2-methylpropanoyl}oxazolidin-2-one 8 and (4S)-4-isopropyl-3-{(2S)-[3-13C]-2-methylpropanoyl}oxazolidin-2-one 11 as a pale yellow oil; [α]D +89.9 (c 5.0 in CH2Cl2); δH (500 MHz) 0.88 [3 H, d, J 7.0, (CH3)2CH], 0.91 [3 H, d, J 7.0, (CH3)2CH], 1.15 (0.2 H, dd, J 127.9 and 6.9, 1113CH3), 1.15 (2.8 H, dd, J 6.9 and 5.2, 8 CHCH313CH3), 1.22 (2.8 H, dd, J 128.3 and 6.9, 813CH3), 1.22 (0.2 H, dd, J 6.9 and 5.0, 11 CHCH313CH3), 2.34 [1 H, septet d, J 7.0 and 4.0, CH(CH3)2], 3.79 (1 H, septet d, J 6.9 and 5.0, CHCH313CH3), 4.20 (1 H, dd, J 9.2 and 3.1, CH2O), 4.27 (1 H, dd, J 9.2 and 8.4, CH2O), and 4.45 (1 H, ddd, J 8.4, 4.0 and 3.1, CHN); δC 14.6 [CH(CH3)2], 17.8 [CH(CH3)2], 18.2 (CHCH313CH3), 19.5 (CHCH313CH3), 28.3 [CH(CH3)2], 32.5 (d, J 34, CHCH313CH3), 58.3 (CHN), 63.2 (CH2O), 153.6 (NCO2) and 177.5 (d, J 2, CON); m/z 200 (M+, 24%, 93% incorporation of carbon-13), 185 (5), 157 (14), 130 (52), 86 (44), 78 (15), 72 (100) and 58 (66).
The above reaction was repeated using (4S)-4-isopropyl-3-{[3-13C]propanoyl}oxazolidin-2-one 10 and iodomethane giving a 13∶1 mixture of (4S)-4-isopropyl-3-{(2S)-[3-13C]-2-methylpropanoyl}oxazolidin-2-one 11 and (4S)-4-isopropyl-3-{(2R)-[3-13C]-2-methylpropanoyl}oxazolidin-2-one 8 as a pale yellow oil; [α]D +84.0 (c 3.5 in CHCl3); δH (500 MHz) 0.88 [3 H, d, J 7.0, (CH3)2CH], 0.91 [3 H, d, J 7.0, (CH3)2CH], 1.15 (2.8 H, dd, J 128.3 and 6.9, 1113CH3), 1.15 [0.2 H, dd, J 6.9 and 5.0, 8 CH3(13CH3)CH], 1.22 (0.2 H, dd, J 127.9 and 6.9, 813CH3), 1.22 [2.8 H, dd, J 6.9 and 5.2, 11 CH3(13CH3)CH], 2.34 [1 H, septet d, J 7.0 and 4.0, CH(CH3)2], 3.79 (1 H, septet d, J 6.9 and 5.0, CHCH313CH3), 4.20 (1 H, dd, J 9.2 and 3.1, CHHO), 4.27 (1 H, dd, J 9.2 and 8.4, CHHO), and 4.45 (1 H, ddd, J 8.4, 4.0 and 3.1, CHN); δC 14.6 [(CH3)2CH], 17.8 [(CH3)2CH], 18.2 (CHCH313CH3), 19.6 (CHCH313CH3), 28.4 [CH(CH3)2], 32.5 (d, J 34.9, CHCH313CH3), 58.3 (CHN), 63.2 (CH2O), 153.6 (NCO2) and 177.5 (d, J 1.5, CON); m/z 200 (M+, 18%, 87% incorporation of carbon-13), 173 (3), 157 (15) 130 (52), 86 (42), 72 (100) and 58 (60).
43,44); δH (270 MHz) 0.89 (3 H, d, J 6.8, CH3CH), 1.30 (3 H, t, J 7.1, OCH2CH3), 2.09 (1 H, m, 4-H), 2.64 (2 H, d, J 6.8, 3-H2), and 4.24 (2 H, q, J 7.1, OCH2CH3); δC (68 MHz) 13.9 (OCH2CH3), 21.3 (quin, J 19, CD3), 22.3 (CH3CH), 23.9 (C-4), 47.7 (C-3), 62.3 (OCH2), 161.3 (C-1), and 194.4 (C-2); m/z 161 (M+, 3%, 97% incorporation of 3 deuterium atoms), 116 (2), 97 (44), 88(44), 85 (50), 71 (72), 60 (39) and 57 (100).
The aqueous solution was adjusted to pH 1–2 with 1 M sulfuric acid and extracted with ether (3 × 125 mL). The combined extracts were dried over sodium sulfate and evaporated to yield (2R)-[3-13C]-2-methylpropanoic acid45 (1.27 g, quantitative) as a colourless liquid; δH1.20 (3 H, dd, J 128.1 and 7.0, 13CH3), 1.20 (3 H, dd, J 7.0 and 5.2, CH3), 2.59 (1 H, septet d, J 7.0 and 4.6, CH) and 5.96 (br s, CO2H); δC 18.7 (13CH3), 33.8 (d, J 34, CH) and 183.4 (CO2H).
A solution of (2R)-[3-13C]-2-methylpropanoic acid (1.16 g, 13.0 mmol) in ether (25 mL) was added dropwise to a stirred slurry of lithium aluminium hydride (0.99 g, 26 mmol) in ether (40 mL) at 0 °C and the resultant mixture stirred at 0 °C for 6 h. Water (6 mL) was added slowly to the mixture and the pH of the resultant grey slurry adjusted to 2–3 with 1 M sulfuric acid. The layers were separated and the aqueous layer extracted with ether (4 × 65 mL). The combined organic extracts were washed with 1 M sodium hydrogen carbonate solution (120 mL), dried over magnesium sulfate and filtered. This solution was distilled (50 °C, atmospheric pressure) to yield, as the residue, a mixture of (2R)-[3-13C]-2-methylpropan-1-ol 13 (861 mg, 88%, calculated from the 1H NMR spectrum) and ether as a colourless liquid (lit. data on deuterated material46), δH (270 MHz) 0.91 (3 H, dd, J 124.8 and 6.7, 13CH3), 0.91 (3 H, dd, J 6.8 and 5.3, CH3), 1.21 (9 H, t, J 7.0, ether CH3), 1.75 (1 H, nonet d, J 6.6 and 3.7, CH), 2.76 (1 H, br s, OH), 3.37 (2 H, dd, J 6.5 and 3.8, CH2) and 3.48 (6 H, q, J 7.0, ether CH2).
A solution of (2R)-[3-13C]-2-methylpropan-1-ol 13 [ca. 625 mg, ca. 8.3 mmol, carried through from the preceding step in ether–deuterochloroform (ca. 3 mL)] and pyridine (1.2 mL, 15 mmol) in carbon tetrachloride (17 mL) was added dropwise over ca. 40 min to a stirred solution of trifluoromethanesulfonic anhydride (2.4 mL, 14 mmol) in carbon tetrachloride (13 mL) at 0 °C. The resultant solution was stirred at 0 °C for a total of 55 min then water (30 mL) was added and the layers separated. The aqueous layer was extracted with dichloromethane (2 × 50 mL), the combined organic extracts dried over magnesium sulfate, filtered and evaporated until just dry, to yield crude (2R)-[3-13C]-2-methylpropyl trifluoromethanesulfonate 14 (1.26 g, ca. 73%) as a cloudy brown oil (lit. data on unlabelled material47), δH (270 MHz) 1.03 (3 H, dd, J 126.4 and 6.7, 13CH3), 1.03 (3 H, dd, J 6.6 and 5.1, CH3), 2.13 (1 H, nonet d, J 6.7 and 4.1, CH) and 4.31 (2 H, ddd, J 6.4, 3.8 and 0.5, CH2).
A solution of 2-ethoxycarbonyl-1,3-dithiane (0.96 mL, 6.1 mmol) in THF (2.5 mL) was added dropwise to n-butyllithium (2.8 mL, 2.44 M solution in hexanes) in THF (5 mL) at −78 °C for 20 min. Crude (2R)-[3-13C]-2-methylpropyl trifluoromethanesulfonate 14 (1.26 g, ca. 6.1 mmol) was added dropwise and the mixture stirred at −78 °C for 15 min then at rt overnight. Sat. sodium hydrogen carbonate solution (50 mL) was added and the mixture extracted with ethyl acetate (5 × 60 mL). The combined organic extracts were washed successively with water (100 mL) and brine (100 mL), dried over magnesium sulfate, filtered and evaporated. Column chromatography of the resultant yellow oil with 1–5% ethyl acetate–light petroleum as eluent yielded pure 2-ethoxycarbonyl-2-{(2R)-[3-13C]-2-methylpropyl}-1,3-dithiane 15 (820 mg, 56%) as a colourless oil; δH (270 MHz) 0.94 (3 H, dd, J 125.1 and 6.4, 13CH3), 0.94 (3 H, dd, J 6.5 and 5.4, CHCH3), 1.33 (3 H, t, J 7.1, OCH2CH3), 1.86 (1 H, dtt, J 14.0, 12.5 and 3.3, SCH2CHHax), 1.94–2.06 (3 H, m, 3-H2 and 4-H), 2.14 (1 H, dtt, J 14.0, 4.5 and 2.5, SCH2CHHeq), 2.65 (2 H, ddd, J 14.5, 4.5 and 3.3, 2 × SCHHeq), 3.26 (2 H, ddd, J 14.5, 12.5 and 2.5, 2 × SCHHax) and 4.24 (2 H, q, J 7.1, CO2CH2CH3); δC 14.1 (CH2CH3), 23.7 (13CH3 and CH3), 24.8 (SCH2CH2), 25.2 (d, J 35, C-4), 27.9 (SCH2), 47.0 (C-3), 53.1 (d, J 3, CS2), 61.8 (OCH2CH3) and 171.4 (CO); followed by mixed fractions containing both 15 (319 mg, 21%) and 2-ethoxycarbonyl-1,3-dithiane, then pure 2-ethoxycarbonyl-1,3-dithiane.
)-crotonyl-1,10-camphorsultam 1949 (1.06 g, 3.5 mmol) in toluene (40 mL) was added slowly to the homocuprate solution (during the addition the reaction mixture turned yellow again) and the resultant mixture was stirred at −78 °C for 6 h. The reaction was quenched at −78 °C with a suspension of sat. ammonium chloride (40 mL) in THF (40 mL) and warmed to rt. The layers were separated and the aqueous layer extracted with ether (3 × 50 mL). The combined organic extracts were washed with sat. ammonium chloride solution (30 mL), dried over magnesium sulfate, filtered and evaporated. Column chromatography of the resultant cloudy white oil yielded a 10∶1 mixture of (1S)-N-{(3S)-[4-13C]-3-methylbutanoyl}-1,10-camphorsultam 20 and (1S)-N-{(3R)-[4-13C]-3-methylbutanoyl}-1,10-camphorsultam (1.0 g, 89%) as a white solid; mp 126–127 °C (lit.,50 unlabelled material 129–130 °C); [α]D
−88.57 (c 1.18 in CHCl3) [lit.,50 unlabelled material −89.16 (c 1.14 in CHCl3)]; δH (400 MHz) 0.95–0.99 (6 H, overlapping signals, 8-H3 or 9-H3 and CHCH3), 0.96 (0.5 H,† dd, J 125.2 and 6.6, 13CH3 minor diastereomer), 0.97 (3 H, s, CH3), 0.97 (2.5 H,† dd, J 125.2 and 6.8, 13CH3 major diastereomer), 1.16 (3 H, s, 8-H3 or 9-H3), 1.32–1.44 (2 H, m, 5-H and 6-H), 1.87–1.96 (3 H, m, 4-H, 5-H and 6-H), 2.08–2.10 (2 H, m, 3-H2), 2.23 (1 H, nonet d, J 6.8 and 3.8, CHCH3), 2.52 (1 H, ddd, J 15.6, 6.8 and 3.4, COCHH), 2.66 (1 H, ddd, J 15.7, 7.1 and 4.6, COCHH), 3.43 (1 H, d, J 13.9, 10-HH), 3.49 (1 H, d, J 13.9, 10-HH) and 3.88 (1 H, t, J 6.4, 2-H); δC (100 MHz) 19.9, 20.8 (C-8 and C-9), 22.3 (slightly enriched CH3), 22.3 (enriched CH3), 25.6 (d, J 35, CHCH3), 26.5, 32.9 (C-5 and C-6), 38.6 (C-3), 44.2 (COCH2), 44.7 (C-4), 47.7, 48.3 (C-1 and C-7), 53.1 (C-10), 65.2 (C-2) and 171.5 (CO); m/z 300 (M+, 300.1574, C1413CH25NO3S requires 300.1589, 3%), 285 (1), 257 (22), 151 (12), 135 (25), 134 (25), 108 (17), 93 (16), 86 (100) and 58 (57).
51 (930 mg, 93%) as a colourless liquid; δH (270 MHz) 0.99 (3 H, dd, J 6.6 and 5.3, CH3), 0.99 (3 H, dd, J 125.2 and 6.6, 13CH3), 2.05–2.17 (1 H, m, CH), 2.21–2.26 (2 H, m, CH2) and 8.85 (1 H, br s, CO2H); δC 22.4 (enriched CH3), 25.5 (d, J 35, CH), 43.2 (CH2) and 179.6 (d, J 3, CO2H) .
A mixture of (S)-[4-13C]-3-methylbutanoic acid 21 (0.96 g, 9.3 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) (2.7 g, 14 mmol) and 4-dimethylaminopyridine (DMAP) (110 mg, 0.9 mmol) in dichloromethane (120 mL) was stirred at 0 °C for 10 min before the solution of (cyanomethylene)triphenylphosphorane 22 was added dropwise over ca. 20 min. The mixture was warmed to rt and stirred overnight. Water (200 mL) was added, the layers separated and the aqueous layer extracted with dichloromethane (2 × 200 mL). The combined organic extracts were washed successively with sat. sodium hydrogen carbonate solution (100 mL) and brine (100 mL), dried over magnesium sulfate, filtered and evaporated. Column chromatography of the resultant yellow oil with 5–10% ethyl acetate in dichloromethane as eluent yielded (S)-[6-13C]-5-methyl-3-oxo-2-triphenylphosphoranylidenehexanenitrile 23 (2.87 g, 80%) as an off-white solid; δH (270 MHz) 0.96 (3 H, dd, J 6.6 and 5.3, CH3), 0.96 (3 H, dd, J 124.7 and 6.6, 13CH3), 2.21 (1 H, nonet d, J 6.8 and 3.7, 5-H), 2.58 (2 H, dd, J 7.1 and 4.3, 4-H2) and 7.46–7.66 (15 H, m, Ph); δC (67.9 MHz) 22.5 (enriched, CH3), 25.8 (d, J 36, C-5), 48.1 (d, J 8, C-4), 49.3 (d, J 126, C-2), 122.6 (d, J 16, C-1), 123.3 (d, J 94, Ph ipso-C), 128.9 (d, J 13, aromatic), 132.8 (d, J 2, aromatic), 133.4 (d, J 10, aromatic) and 196.7 (C-2).
Ozone was passed through a stirred solution of (5S)-[6-13C]-5-methyl-3-oxo-2-triphenylphosphoranylidenehexanenitrile 23 (2.87 g, 7.4 mmol) in dichloromethane (60 mL) and methanol (30 mL) at −78 °C until the solution remained blue (ca. 2 h). The solution was then purged with oxygen, at −78 °C, until colourless and finally warmed to rt. The reaction mixture was evaporated to dryness and the resultant yellow solid purified by column chromatography with 25% light petroleum in chloroform as eluent to yield methyl (4S)-[5-13C]-4-methyl-3-oxopentanoate 24 (810 mg, 76%) as a colourless liquid (lit. data on unlabelled material53); δH (270 MHz) 0.97 (3 H, dd, J 6.6 and 5.3, CH3), 0.97 (3 H, dd, J 125.4 and 6.7, 13CH3), 2.20 (1 H, nonet d, J 6.7 and 3.8, 4-H), 2.73 (2 H, dd, J 6.8 and 4.2, 3-H2) and 3.87 (3 H, s, CO2CH3); δC (67.9 MHz) 22.4 (enriched, CH3), 24.2 (d, J 35, C-4), 47.9 (C-3), 52.8 (OCH3), 161.7 (C-1) and 193.9 (C-2).
![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )-Crotonyl]-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 27..
Crotonyl chloride (2.0 mL, 21 mmol) was added to (1R,2S,3R)-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 26 (1.19 g, 2.9 mmol) in carbon tetrachloride (100 mL) and the solution heated under reflux in the presence of 4 Å molecular sieves overnight. After the reaction mixture had been cooled to rt, the carbon tetrachloride was removed in vacuo. The residual oil was dissolved in ethyl acetate (20 mL) and washed with sat. sodium hydrogen carbonate solution (30 mL). The aqueous layer was extracted with ethyl acetate (30 mL). The combined organic extracts were dried over sodium sulfate, filtered and evaporated. The crude product was recrystallised from dichloromethane and light petroleum to give (1R,2S,3R)-2-[(E)-crotonyl]-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 27 (1.12 g, 81%) as a white solid, mp 166–167 °C (data not previously reported33b); [α]D +1.51 (c 1.96 in CHCl3); νmax (Nujol)/cm−1 1719 (CO), 1660 (C
)-Crotonyl]-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 27..
Crotonyl chloride (2.0 mL, 21 mmol) was added to (1R,2S,3R)-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 26 (1.19 g, 2.9 mmol) in carbon tetrachloride (100 mL) and the solution heated under reflux in the presence of 4 Å molecular sieves overnight. After the reaction mixture had been cooled to rt, the carbon tetrachloride was removed in vacuo. The residual oil was dissolved in ethyl acetate (20 mL) and washed with sat. sodium hydrogen carbonate solution (30 mL). The aqueous layer was extracted with ethyl acetate (30 mL). The combined organic extracts were dried over sodium sulfate, filtered and evaporated. The crude product was recrystallised from dichloromethane and light petroleum to give (1R,2S,3R)-2-[(E)-crotonyl]-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 27 (1.12 g, 81%) as a white solid, mp 166–167 °C (data not previously reported33b); [α]D +1.51 (c 1.96 in CHCl3); νmax (Nujol)/cm−1 1719 (CO), 1660 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C), 1607 and 1593 (CC, Ar), δH (300MHz) 0.65, 0.78 and 0.99 (3 × 3 H, 3 × s, 8-H3, 9-H3 and 10-H3), 1.08, 1.35, 1.53, 1.72 (each 1 H, m, 5-H2 and 6-H2), 1.93 (3 H, dd, J 7, 2, CHCH3), 2.07 (3 H, br s, Ar-CH3), 2.16 (1 H, m, 4-H), 2.26 (3 H, br s, Ar-CH3), 3.82 (1 H, d, J 7, 3-H), 5.21 (1 H, d, J 7, 2-H), 5.91 (1 H, dq, J 15.5, 2, COCH), 6.03 (1 H, br s, Ar-H), 6.83 (2 H, br s, Ar-H), 7.03 (1 H, dq, J 15.5, 7, COCHCH), 7.44 (5 H, m, Ar-H); δC 11.2, 20.8 and 21.2 (C-8, C-9 and C-10), 18.1 (CHCH3), 21.1 (Ar-CH3), 27.6 and 32.0 (C-5 and C-6), 47.4 and 50.2 (C-1 and C-7), 48.6 (C-4), 67.3 (C-3), 80.4 (C-2), 123.1 (COCH), 128.1, 128.3, 129.2, 132.4, 137.0 and 139.2 (C-Ar), 144.1 (COCHCH) and 165.4 (CO); m/z (CI) 482 [(M + H)+, 482.2372, C28H36NO4S requires 482.2365, 12%], 396 (74), 341 (100), 254 (70), 226 (8).
)-crotonyl]-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 26 (204 mg, 0.42 mmol) in ether (8 mL) was added dropwise and the reaction mixture stirred at −78 °C for 1 h, −40 °C for 3 h and finally −20 °C for 3 h. The reaction was quenched at −20 °C with sat. ammonium chloride solution (50 mL), before warming to rt. The layers were separated and the aqueous layer extracted with ether (3 × 50 mL). The combined organic extracts were washed with sat. ammonium chloride solution (50 mL), dried over sodium sulfate, filtered and evaporated to yield a yellow solid. Purification by column chromatography, eluting with 0–10% ethyl acetate in light petroleum gave (1R,2S,3R)-2-{(3R)-[4-13C]-3-methylbutanoyl}-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 28 as a white solid (165 mg, 79%); mp 173–174 °C; [α]D
−3.8 (c 1.05 in CHCl3); νmax (CHCl3)/cm−1 1733 (CO); δH [500 MHz; (CD3)2CO] 0.63, 0.79 and 0.94 (3 × 3 H, 3 × s, 8-H3, 9-H3 and 10-H3), 0.99 (3 H, dd, J 125, 6.5, 13CH3), 1.00 (3 H, dd, J 6.4, 5.3, CHCH3), 1.22, 1.28, 1.54 and 1.70 (each 1 H, each m, 5-H2 and 6-H2), 2.04–2.34 (10 H, m, 2 × Ar-CH3, 4-H, and COCH2CH), 4.00 (1 H, d, J 7.0, 3-H), 5.17 (1 H, d, J 7.0, 2-H), 6.05 (1 H, br s, Ar-H), 6.90 (1 H, br s, Ar-H), 6.95 (1H, br s, Ar-H), 7.43–7.64 (5 H, m, Ar-H); δC [75MHz, (CD3)2CO] 11.9, 21.1, 21.2, 21.8 and 25.5 (C-8, C-9, C-10 and 2 × Ar-CH3), 22.9 (13CH3), 23.0 (CHCH3), 28.0 and 32.8 (C-5 and C-6), 44.2 (COCH2), 48.0 and 50.7 (C-1 and C-7), 49.4 (C-4), 68.0 (C-3), 81.6 (C-2), 129.0, 129.2, 129.8, 133.5, 138.5 and 140.3 (C-Ar), 172.0 (CO); m/z (CI) 499 [(MH)+, 499.2714, C2813CH40NO4S requires 499.2712, 13%], 396 (50), 358 (100), 254 (90), 85 (36).
C), 1607 and 1593 (CC, Ar), δH (300MHz) 0.65, 0.78 and 0.99 (3 × 3 H, 3 × s, 8-H3, 9-H3 and 10-H3), 1.08, 1.35, 1.53, 1.72 (each 1 H, m, 5-H2 and 6-H2), 1.93 (3 H, dd, J 7, 2, CHCH3), 2.07 (3 H, br s, Ar-CH3), 2.16 (1 H, m, 4-H), 2.26 (3 H, br s, Ar-CH3), 3.82 (1 H, d, J 7, 3-H), 5.21 (1 H, d, J 7, 2-H), 5.91 (1 H, dq, J 15.5, 2, COCH), 6.03 (1 H, br s, Ar-H), 6.83 (2 H, br s, Ar-H), 7.03 (1 H, dq, J 15.5, 7, COCHCH), 7.44 (5 H, m, Ar-H); δC 11.2, 20.8 and 21.2 (C-8, C-9 and C-10), 18.1 (CHCH3), 21.1 (Ar-CH3), 27.6 and 32.0 (C-5 and C-6), 47.4 and 50.2 (C-1 and C-7), 48.6 (C-4), 67.3 (C-3), 80.4 (C-2), 123.1 (COCH), 128.1, 128.3, 129.2, 132.4, 137.0 and 139.2 (C-Ar), 144.1 (COCHCH) and 165.4 (CO); m/z (CI) 482 [(M + H)+, 482.2372, C28H36NO4S requires 482.2365, 12%], 396 (74), 341 (100), 254 (70), 226 (8).
)-crotonyl]-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 26 (204 mg, 0.42 mmol) in ether (8 mL) was added dropwise and the reaction mixture stirred at −78 °C for 1 h, −40 °C for 3 h and finally −20 °C for 3 h. The reaction was quenched at −20 °C with sat. ammonium chloride solution (50 mL), before warming to rt. The layers were separated and the aqueous layer extracted with ether (3 × 50 mL). The combined organic extracts were washed with sat. ammonium chloride solution (50 mL), dried over sodium sulfate, filtered and evaporated to yield a yellow solid. Purification by column chromatography, eluting with 0–10% ethyl acetate in light petroleum gave (1R,2S,3R)-2-{(3R)-[4-13C]-3-methylbutanoyl}-3-[N-phenylsulfonyl-N-(3,5-dimethylphenyl)amino]bornan-2-ol 28 as a white solid (165 mg, 79%); mp 173–174 °C; [α]D
−3.8 (c 1.05 in CHCl3); νmax (CHCl3)/cm−1 1733 (CO); δH [500 MHz; (CD3)2CO] 0.63, 0.79 and 0.94 (3 × 3 H, 3 × s, 8-H3, 9-H3 and 10-H3), 0.99 (3 H, dd, J 125, 6.5, 13CH3), 1.00 (3 H, dd, J 6.4, 5.3, CHCH3), 1.22, 1.28, 1.54 and 1.70 (each 1 H, each m, 5-H2 and 6-H2), 2.04–2.34 (10 H, m, 2 × Ar-CH3, 4-H, and COCH2CH), 4.00 (1 H, d, J 7.0, 3-H), 5.17 (1 H, d, J 7.0, 2-H), 6.05 (1 H, br s, Ar-H), 6.90 (1 H, br s, Ar-H), 6.95 (1H, br s, Ar-H), 7.43–7.64 (5 H, m, Ar-H); δC [75MHz, (CD3)2CO] 11.9, 21.1, 21.2, 21.8 and 25.5 (C-8, C-9, C-10 and 2 × Ar-CH3), 22.9 (13CH3), 23.0 (CHCH3), 28.0 and 32.8 (C-5 and C-6), 44.2 (COCH2), 48.0 and 50.7 (C-1 and C-7), 49.4 (C-4), 68.0 (C-3), 81.6 (C-2), 129.0, 129.2, 129.8, 133.5, 138.5 and 140.3 (C-Ar), 172.0 (CO); m/z (CI) 499 [(MH)+, 499.2714, C2813CH40NO4S requires 499.2712, 13%], 396 (50), 358 (100), 254 (90), 85 (36).
Acknowledgements
We thank EPSRC for CASE studentships (RAH and NMK) and a postdoctoral fellowship (MDF), University of Bristol for a Scholarship (AS) and Amersham International (NMK) and Zeneca Pharmaceuticals (RAH) for financial support. We are grateful to Drs R. G. Reid and P. Winton for valuable discussions.
References
- N. M. Kelly, A. Sutherland and C. L. Willis, Nat. Prod. Rep., 1997, 14, 205 RSC.
- F. J. Winkler, K. Kuhnl, R. Medina, R. Schwarz-Koske and H. L. Schmidt, Isot. Environ. Health Stud., 1995, 31, 161 Search PubMed.
- D. W. Young, Top. Stereochem., 1994, 21, 381 Search PubMed.
- N. Sitachitta, J. Rossi, M. A. Roberts, W. H. Gerwick, M. D. Fletcher and C. L. Willis, J. Am. Chem. Soc., 1998, 120, 7131 CrossRef CAS.
- R. Cardillo, C. Fuganti, D. Ghiringhelli, P. Grasselli and G. Gatti, J. Chem. Soc., Chem. Commun., 1977, 474 RSC.
- C. Fuganti, P. Grasselli and G. Pedrocchi-Fantoni, Tetrahedron Lett., 1979, 20, 2453 CrossRef.
- P. Anastasis, I. Freer, K. H. Overton, D. Picken, D. S. Rycroft and S. B. Singh, J. Chem. Soc., Perkin Trans. 1, 1987, 2427 RSC.
- D. W. Young, Chem. Soc. Rev., 1994, 23, 119 RSC.
- M. P. Crump, J. Crosby, C. E. Dempsey, J. A. Parkinson, M. Murray, D. A. Hopwood and T. J. Simpson, Biochemistry, 1997, 36, 6000 CrossRef CAS.
- S. R. Sylvester and C. M. Stevens, Biochemistry, 1979, 18, 4529 CrossRef CAS; S. R. Sylvester, S. Y. Lan and C. M. Stevens, Biochemistry, 1981, 20, 5609 CrossRef CAS.
- D. J. Aberhart and B. H. Weiller, J. Labelled Compd. Radiopharm., 1983, 20, 663 CAS.
- R. K. Hill, C. Abacherli and S. Hagishita, Can. J. Chem., 1994, 72, 110 CAS.
- J. C. Shuttuck and J. Meinwald, Tetrahedron Lett., 1997, 38, 8461 CrossRef CAS.
- R. A. August, J. A. Khan, C. M. Moody and D. W. Young, Tetrahedron Lett., 1992, 33, 4617 CrossRef CAS; R. A. August, J. A. Khan, C. M. Moody and D. W. Young, J. Chem. Soc., Perkin Trans. 1, 1996, 507 RSC.
- M. Oba, T. Terauchi, A. Miyakawa, H. Kamo and K. Nishiyama, Tetrahedron Lett., 1998, 39, 1595 CrossRef CAS.
- N. M. Kelly, R. G. Reid, C. L. Willis and P. L. Winton, Tetrahedron Lett., 1995, 36, 8315 CrossRef CAS.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127 CrossRef CAS; D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS.
- For reviews of chiral auxiliaries derived from amino acids, including Evans’ auxiliaries see: (a) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS; (b) A. Studer, Synthesis, 1996, 793 CrossRef CAS.
- For reviews of α-keto esters and acids see: (a) A. J. L. Cooper, J. Z. Ginos and A. Meister, Chem. Rev., 1983, 83, 321 CrossRef CAS; (b) L. Kovács, Recl. Trav. Chim. Pays-Bas, 1993, 112, 471 Search PubMed.
- J. Buckingham , S. M. Donaghy , J. I. G. Cadogan , R. A. Raphael and C. W. Rees , Dictionary of Organic Compounds, Chapman and Hall, New York, 1982. Search PubMed.
- Z. Shaked and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 7104 CrossRef CAS.
- N. M. Kelly, R. G. Reid, C. L. Willis and P. L. Winton, Tetrahedron Lett., 1996, 37, 1517 CrossRef CAS.
- M. J. Garson and J. Staunton, Chem. Soc. Rev., 1979, 8, 539 RSC.
- N. M. Kelly, B. C. O’Neill, J. Probert, G. Reid, R. Stephen, T. Wang, C. L. Willis and P. Winton, Tetrahedron Lett., 1994, 35, 6533 CrossRef CAS.
- E. L. Eliel and A. A. Hartmann, J. Org. Chem., 1972, 37, 505 CrossRef CAS; G. S. Bates , in Encyclopedia of Reagents for Organic Synthesis, ed. L. A. Paquette, Wiley, Chichester, 1995, vol. 4, p. 2623. Search PubMed.
- D. A. Evans, T. C. Britton and J. A. Ellman, Tetrahedron Lett., 1987, 28, 6141 CrossRef CAS.
- M. Imoto, S. Kusumoto and T. Shiba, Tetrahedron Lett., 1987, 28, 6235 CrossRef CAS.
- E. J. Corey and B. W. Erickson, J. Org. Chem., 1971, 36, 3553 CrossRef CAS.
- For reviews of stereoselective conjugate additions see for example: (a) B. E. Rossiter and N. M. Swingle, Chem. Rev., 1992, 92, 771 CrossRef CAS; (b) A. Alexakis , in Organocopper Reagents: A Practical Approach, ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994, p. 159; Search PubMed; (c) P. Perlmutter , Conjugate Addition Reactions in Organic Synthesis, Pergamon, Oxford, 1992. Search PubMed.
- G. Pourcelot, J. Aubouet, A. Caspar and P. Cresson, J. Organomet. Chem., 1987, 328, C43 CrossRef CAS.
- (a) Y. Han and V. J. Hruby, Tetrahedron Lett., 1997, 38, 7317 CrossRef CAS; (b) W. Yuan and V. J. Hruby, Tetrahedron Lett., 1997, 38, 3853 CrossRef CAS; (c) X. Qian, K. C. Russell, L. W. Boteju and V. J. Hruby, Tetrahedron, 1995, 51, 1033 CrossRef CASand references therein..
- (a) D. R. Williams, W. S. Kissel and J. J. Li, Tetrahedron Lett., 1998, 39, 8593 CrossRef CAS; (b) D. R. Williams and J. J. Li, Tetrahedron Lett., 1994, 35, 5113 CrossRef CAS.
- (a) W. Oppolzer, R. J. Mills, W. Pachinger and T. Stevenson, Helv. Chim. Acta, 1986, 69, 1542 CrossRef CAS; (b) M. Vandewalle, J. Van der Eycken, W. Oppolzer and C. Vullioud, Tetrahedron, 1986, 42, 4035 CrossRef CAS; (c) W. Oppolzer, G. Poli, A. J. Kingma, C. Starkemann and G. Bernardinelli, Helv. Chim. Acta, 1987, 70, 2201 CrossRef CAS; (d) W. Oppolzer, A. J. Kingma and G. Poli, Tetrahedron, 1989, 45, 479 CrossRef CAS; (e) W. Oppolzer and A. J. Kingma, Helv. Chim. Acta, 1989, 72, 1337 CrossRef CAS; (f) W. Oppolzer, Pure Appl. Chem., 1990, 62, 1241 CrossRef CAS; (g) W. Oppolzer, Tetrahedron, 1987, 43, 1969 CrossRef CAS.
- H. H. Wasserman and W.-B. Ho, J. Org. Chem., 1994, 59, 4364 CrossRef CAS.
- (a) G. Helmchen and G. Wegner, Tetrahedron Lett., 1985, 26, 6051 CrossRef CAS; (b) G. Helmchen and G. Wegner, Tetrahedron Lett., 1985, 26, 6047 CrossRef CAS; (c) E. Urban, G. Riehs and G. Knühl, Tetrahedron Lett., 1995, 36, 4773 CrossRef CAS; (d) E. Urban, G. Knühl and G. Helmchen, Tetrahedron, 1996, 52, 971 CrossRef CAS.
- Y. Yamamoto and K. Maruyama, J. Am. Chem. Soc., 1978, 100, 3240 CrossRef CAS; Y. Yamamoto, S. Yamamoto, H. Yatagai, Y. Ishihara and K. Maruyama, J. Org. Chem., 1982, 47, 119 CrossRef CAS.
- T. Ibuka and Y. Yamamoto , in Organocopper Reagents: A Practical Approach, ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994, p. 143. Search PubMed.
- W. Oppolzer, R. Moretti, T. Godel, A. Meunier and H. Löher, Tetrahedron Lett., 1983, 24, 4971 CrossRef CAS.
- J. A. MacRitchie, A. Silcock and C. L. Willis, Tetrahedron: Asymmetry, 1997, 8, 3895 CrossRef CAS.
- R. J. K. Taylor and G. Casy , in Organocopper Reagents: A Practical Approach, ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994, p. 27. Search PubMed.
- G. B. Kauffman and L. A. Teter, Inorg. Synth., 1963, 7, 9 CAS.
- A. S. Kende, K. Kawamura and M. J. Orwat, Tetrahedron Lett., 1989, 30, 5821 CrossRef CAS.
- J. Singh, T. P. Kissick and R. H. Mueller, Org. Prep. Proced. Int., 1989, 21, 501 Search PubMed.
- J. H. Babler, C. J. Marcuccilli and J. E. Oblong, Synth. Commun., 1990, 20, 1831 CAS.
- K. A. Reynolds, D. O’Hagan, D. Gani and J. A. Robinson, J. Chem. Soc., Perkin Trans. 1, 1988, 3195 RSC.
- J. K. MacDougall, M. C. Simpson and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1994, 3061 RSC.
- M. F. Salomon, R. G. Salomon and R. D. Gleim, J. Org. Chem., 1976, 41, 3983 CrossRef CAS.
- Y. Shimojima, H. Hayashi, T. Ooka and M. Shibukawa, Tetrahedron, 1984, 40, 2519 CrossRef CAS.
- W. Oppolzer and J.-P. Barras, Helv. Chim. Acta, 1987, 70, 1666 CrossRef CAS.
- W. Oppolzer, O. Tamura and J. Deerberg, Helv. Chim. Acta, 1992, 75, 1965 CrossRef CAS.
- J. E. Baldwin, J. Lologer, W. Rastetter, N. Neuss, L. L. Huckstep and N. De La Higuera, J. Am. Chem. Soc., 1973, 95, 3796 CrossRef CAS.
- S. Trippett and D. M. Walker, J. Chem. Soc., 1959, 3874 RSC; G. P. Schiemenz and H. Engelhard, Chem. Ber., 1961, 94, 578 Search PubMed.
- H. Poisel, Chem. Ber., 1978, 111, 3136 Search PubMed.
- W. A. H. Huffman and A. W. Ingersoll, J. Am. Chem. Soc., 1951, 73, 3366 CrossRef CAS.
Footnote |
| † Integral determined from decoupling experiment (irradiation at δH 2.23). |
| This journal is © The Royal Society of Chemistry 2000 |