An efficient semisynthesis of 7-deoxypaclitaxel from taxine
Radomir N.
Saičić
a,
Radomir N.
Saičić
*a and
Radomir
Matović
b
aFaculty of Chemistry, University of Belgrade, Studentski trg 16, P.O. Box 158, YU-11001, Belgrade, Yugoslavia. E-mail: rsaicic@chem.bg.ac.yu.
bICTM, Center for Chemistry, Njegoševa 12, 11000, Belgrade, Yugoslavia
First published on 27th January 2000
Abstract
A semisynthesis of 7-deoxypaclitaxel 4 is described, starting from taxine 6 – the most abundant naturally occurring taxane diterpene fraction. A key step in this transformation is a tandem reaction: stereoselective osmylation of cinnamic ester 14/intramolecularly assisted methanolysis of 16, which gives the key intermediate 5, along with the optically pure ester 17 – a precursor for the synthesis of the paclitaxel side-chain. In this way, the cinnamoyltaxicine 9 is converted into 7-deoxybaccatin III 25 in 11 steps, and in 15% overall yield.
A naturally occurring diterpenoid Taxol
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ® (Paclitaxel) 1 (Scheme 1), isolated from the bark of the Pacific yew (Taxus brevifolia),1 has attracted intense interest from the scientific community, owing to its great potential in the successful treatment of many types of cancer, unusual mode of antimitotic action, and complex molecular architecture.2 Although six total syntheses of Paclitaxel have been reported,3 the structural complexity of the compound makes it elusive for an economical total synthesis. For its commercial production, which was once based solely on extraction from the bark of Taxus brevifolia, which destroys the tree, and arguably threatened the very slow growing yew population, there is now available a useful alternative, which is semisynthesis from 10-O-deacetylbaccatin III (10-DAB) 3, which is in turn isolated, most notably, from the renewable leaves of the European yew, Taxus baccata.4 This method, more environmentally friendly, gave rise to a novel, non-natural taxoid Taxotere® (Docetaxel) 2, which is comparable to, if not better than, Paclitaxel in terms of therapeutic indications and efficiency.5 However, 10-DAB is not the most abundant secondary metabolite of the yew tree: its variable content in the needles of Taxus baccata ranges from 0.01 to 1 g per kg of leaves.6 In addition, the isolation of pure 10-DAB involves somewhat tedious purification by multiple-column chromatography. In contrast, a mixture of alkaloids collectively referred to as ‘Taxine’
6 can be obtained by a simple extraction procedure in yields of 7–12 g kg−1,7 the two major constituents of this fraction (≈35%) being taxine B 6a and isotaxine B 6b.8 Therefore, the development of a procedure that would allow for the efficient use of this starting material for the preparation of biologically active paclitaxel analogues would be of considerable interest.
® (Paclitaxel) 1 (Scheme 1), isolated from the bark of the Pacific yew (Taxus brevifolia),1 has attracted intense interest from the scientific community, owing to its great potential in the successful treatment of many types of cancer, unusual mode of antimitotic action, and complex molecular architecture.2 Although six total syntheses of Paclitaxel have been reported,3 the structural complexity of the compound makes it elusive for an economical total synthesis. For its commercial production, which was once based solely on extraction from the bark of Taxus brevifolia, which destroys the tree, and arguably threatened the very slow growing yew population, there is now available a useful alternative, which is semisynthesis from 10-O-deacetylbaccatin III (10-DAB) 3, which is in turn isolated, most notably, from the renewable leaves of the European yew, Taxus baccata.4 This method, more environmentally friendly, gave rise to a novel, non-natural taxoid Taxotere® (Docetaxel) 2, which is comparable to, if not better than, Paclitaxel in terms of therapeutic indications and efficiency.5 However, 10-DAB is not the most abundant secondary metabolite of the yew tree: its variable content in the needles of Taxus baccata ranges from 0.01 to 1 g per kg of leaves.6 In addition, the isolation of pure 10-DAB involves somewhat tedious purification by multiple-column chromatography. In contrast, a mixture of alkaloids collectively referred to as ‘Taxine’
6 can be obtained by a simple extraction procedure in yields of 7–12 g kg−1,7 the two major constituents of this fraction (≈35%) being taxine B 6a and isotaxine B 6b.8 Therefore, the development of a procedure that would allow for the efficient use of this starting material for the preparation of biologically active paclitaxel analogues would be of considerable interest.
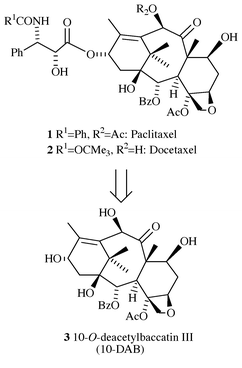 | ||
| Scheme 1 Paclitaxel, Docetaxel, and their common precursor 10-DAB. | ||
Structure–activity relationship (SAR) studies on paclitaxel derivatives have shown that a hydroxy group at position 7 is not essential for biological activity. In fact, reports on 7-deoxypaclitaxel 4, obtained by radical deoxygenation of paclitaxel, or of a baccatin derivative, showed this compound to be of comparable, or even superior, cytotoxic activity with respect to paclitaxel.9 Its structural congruence with taxine B stimulated several groups to investigate the possibility of a semisynthetic approach to 7-deoxypaclitaxel 4, starting from taxine 6.10 As the preparative separation of taxine alkaloids is difficult, the crude taxine mixture was hydrolysed in order to obtain a well defined starting material, i.e. tetraol 7. However, the differentiation between hydroxy groups in tetraol 7 required extensive use of protective groups, leading to long synthetic sequences, and lowering considerably the overall yield.11 Recently, we reported an efficient procedure for the conversion of taxine into 7-deoxypaclitaxel, which in principle could give access not only to the title compound, but also to other derivatives suitable for further SAR studies.12 In this paper we present the results of our study giving full experimental details, together with some observations on the reactivity of the intermediates.
Results and discussion
Our bi-directional retrosynthetic analysis of 7-deoxypaclitaxel is displayed in Scheme 2. While the envisaged transformation of the key intermediate 5 into 7-deoxypaclitaxel 4 relied on protocols previously described on structurally related systems, a new and efficient solution for the retrosynthetic move 5 ⇒ 6 was needed. Two issues had to be addressed: (a) conversion of the taxine mixture 6a–c into a well defined, suitable starting compound, and (b) high-yield transformation of the allylic Winterstein ester fragment (C4–C5–C20) of taxine 6 into the 4,5,20-triol structural subunit in 5, avoiding long synthetic sequences and unnecessary protective group manipulations.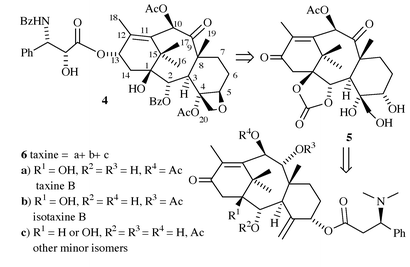 | ||
| Scheme 2 Retrosynthetic analysis of 7-deoxypaclitaxel. | ||
We first re-examined the possibility of exploiting the favourable arrangement of functional groups in the major constituent of taxine, i.e. taxine B 6a (Scheme 3). Quaternization of crude taxine, followed by DBU-induced elimination under anhydrous conditions, afforded a mixture of 9- and 10-O-acetyl-5-O-cinnamoyltaxicine I (8 and 9),13 which could be separated by flash chromatography on a SiO2 column, and were isolated in yields of 1 and 1.7 g kg−1 of needles, respectively. Some time ago, it was reported that 6a and 6b interconvert on storage on TLC silica plates;7c,13 this finding prompted us to examine the possibility of preparative isomerization of 8 into 9. After some experimentation, we found that 8 can be isomerized into the required 10-O-acetyl derivative 9 by treatment with methanolic KOAc (43% yield at 70% conversion, without optimization), to afford a total of 2 g of 10-O-acetyl-5-O-cinnamoyltaxicine I 9 per 1 kg of dry leaves. Later on, we found it advantageous to perform the separation of regiomeric acetates at the level of carbonates 10 and 11, where 11 could be isomerized into 10 on treatment with methanolic KOAc. Alternatively, the mixture of carbonates 10 and 11 can be oxidized with Jones reagent to ketones 14 and 15, which could be easily separated by dry-flash chromatography. We hoped that 11 could be converted into 14 by the following sequence of reactions: a) protection of 11 as the O-ethoxyethyl derivative 12; b) hydrolysis of C-9 acetate in 12 to give alcohol 13, and c) direct oxidation of 13 into the required ketone 14 [eqn. (1)]. These attempts failed, however, as the C-9 acetate in 12 proved resistant towards hydrolysis even under conditions where the cyclic carbonate was cleaved.
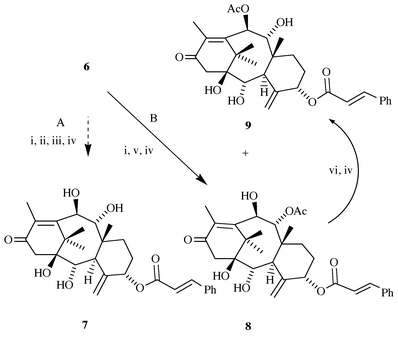 | ||
| Scheme 3 Transformation of taxine into suitable precursor for 7-deoxypaclitaxel synthesis. (A) previous work (ref. 10); (B) this work. Reagents and conditions: (i) MeI, THF, rt, 5 h; (ii) K2CO3, H2O, EtOH, rt, 3 h; (iii) NaOMe, MeOH, 0 °C, 16 h; (iv) column chromatography on SiO2; (v) DBU, CHCl3, rt, 1.5 h; (vi) 10% KOAc in MeOH, rt, 3 h. | ||
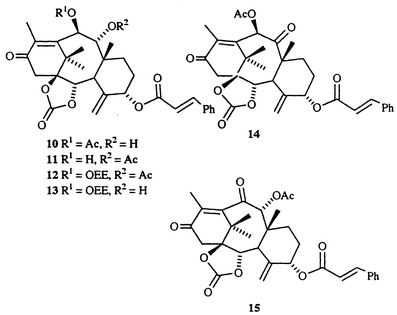
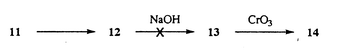
| (1) |
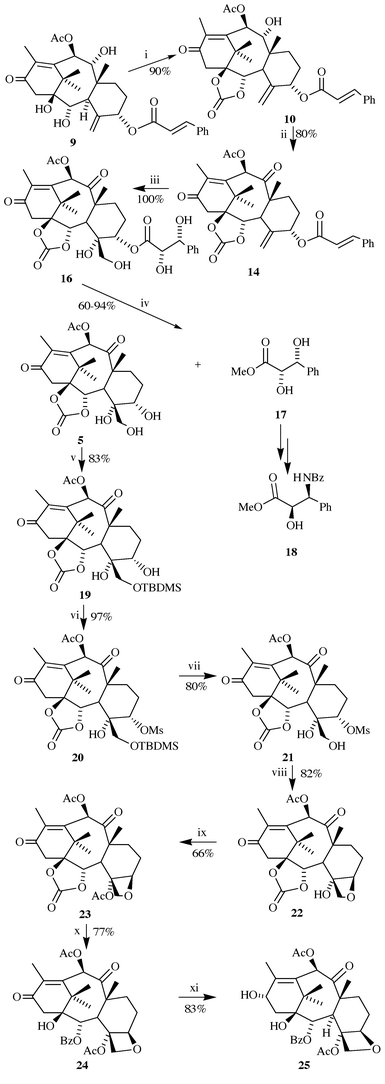
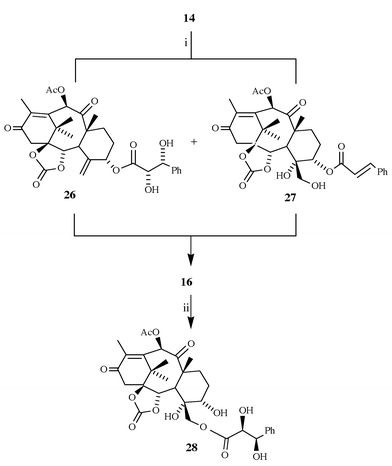
With the suitable starting compound 9 in hand, the synthesis of 7-deoxybaccatin 25 proceeded as displayed in Scheme 4. Treatment of 9 with phosgene, followed by hydrolytic work-up, furnished the cyclic carbonate 10 (90%), which was oxidized to diketone 14 (Dess–Martin periodinane: 80%; PCC: 75%; Jones reagent: 50%), thus establishing the final functionalization of the ‘upper’ part of the molecule. Elaboration of the oxetane ring was envisaged to proceed via triol 5. For that purpose a method for selective removal of the cinnamoyl chain was needed, as the simultaneous cleavage of the C-10 acetate (before, or after, the transformation of the C4–C20 double bond into a vicinal diol) would create a system with two secondary hydroxy groups that might be difficult to differentiate, and which would certainly require additional protective steps. To this end, allylic cinnamate 14 was oxidized with OsO4–NMO, as it was anticipated that intramolecular hydrogen bonding in 16 should activate the dihydroxyphenylpropionate ester towards hydrolysis under very mild conditions; in this way, both the desired functional transformation of the taxane core and the activation of the ‘side-chain’ would be accomplished in a single step. Although some concern existed about the stereochemical outcome of the osmylation, to our pleasure the reaction proceeded with complete stereoselectivity, and in quantitative yield. TLC monitoring of the reaction mixture indicated that the reaction proceeded via two intermediates of close polarity (major: less polar; minor: more polar), which, during the course of the reaction, converged to a single product 16. When the hydroxylation was interrupted before completion, these intermediates could be isolated and identified as diols 26 and 27 (Scheme 5). However, this bifurcate reactivity does not compromise diastereoselectivity, as a single isomer 16 is obtained. Whether intramolecular assistance is included in the reaction mechanism or not remains unclear.
 | ||
| Scheme 4 Reagents and conditions: (i) COCl2 (20 equiv.), CH2Cl2, 0 °C, 20 min; then Et2O, H2O, imidazole (cat.), 0 °C, 30 min; (ii) Dess–Martin periodinane (2 equiv.), CH2Cl2, CF3CO2H (cat.), rt, 12 h; (iii) OsO4 (cat.), NMO, THF, H2O, rt, 4 h; (iv) 10% KOAc in MeOH, reflux, 30 min; (v) TBDMSCl, Et3N, DMAP (cat.), CH2Cl2, rt, 24 h; (vi) MsCl, Py, 0 °C to rt, 24 h; (vii) 7% HF in CH3CN, rt, 7 h; (viii) iPr2NEt (7 equiv.), toluene, reflux, 39 h; (ix) Ac2O (7 equiv.), DMAP (14 equiv.), CH2Cl2, rt, 4 h; (x) PhLi (10 equiv.), THF, −78 °C, 0.5 h; then Ac2O, DMAP, CH2Cl2, rt, 1 h; (xi) NaBH4 (excess), MeOH, rt, 3 h. | ||
 | ||
| Scheme 5 Reagents and conditions: (i) OsO4 (cat.), NMO, THF, H2O, rt; (ii) KOAc, 18-cr-6 (cat.), CH2Cl2, rt, 15 h. | ||
The conversion of 14 to 16 brought about the expected modification in the side-chain ester reactivity, as indicated by its proclivity towards spontaneous migration of the ester side-chain from O-5 to O-20 on storage in solution (MeOH, THF) at room temperature; on treatment with KOAc in CH2Cl2, in the presence of a catalytic amount of 18-crown-6, tetraol 16 was converted into 28 quantitatively. Submitting 16 to K2CO3 or NaHCO3 in aq. methanol at 0 °C induced very rapid hydrolysis of the dihydroxypropanoate ester, but hydrolysis of the C-10 acetate also occurred under these conditions. Eventually, refluxing of 16 with methanolic KOAc afforded the desired triol 5 in 94% yield.†
Stereochemical identity of 5 was verified by an NOE-difference experiment on its C-20 acetate 29 (in 1H NMR spectrum of 5 some signals overlapped) which confirmed the proposed (i.e., correct) configuration at C-4 (Fig. 1). Optically pure (2S,3R)-(−)-(methyl 2,3-dihydroxy-3-phenylpropanoate) 17 was isolated as the side product of this reaction, and further transformed into the paclitaxel side-chain 18 according to the previously published procedure.14 Silylation of the primary alcohol in 5 and mesylation of 5-OH were accomplished in the usual way (80% over two steps), but TBAF- induced deprotection of 20 gave 21 in only 40% yield, indicating its instability under basic conditions, and possible complications in the cyclization step. A higher yield of 21 was obtained when the deprotection was carried out under acidic conditions (HF–CH3CN, 80%). Tetrabutylammonium acetate in refluxing butanone (‘standard’ reagent for the construction of the oxetane ring on structurally similar systems)10a caused the rapid decomposition of 21, as did many other reagents.‡ After considerable experimentation it was found that the oxetane-ring closure could be accomplished with Hunig’s base in refluxing toluene (82%). Acetylation of tertiary alcohol 22 afforded 23 in moderate yield (66%). Installation of the requisite 1-hydroxy, 2-benzoate functionalities, and re-acetylation of the secondary alcohol at C-10 were performed following a known protocol,15 furnishing 24 in 77% yield. Finally, reduction of enone 24 with NaBH4 gave rise to 7-deoxybaccatin III 25 (83% at 85% conversion),9b,c,10d thus completing the synthesis of this compound from 10-O-acetyl-5-O-cinnamoyltaxicine I 9 in 15% overall yield (unoptimized).
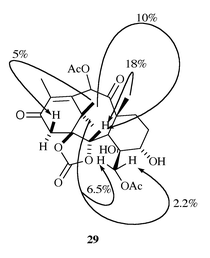 | ||
| Fig. 1 NOE correlations in 29. | ||
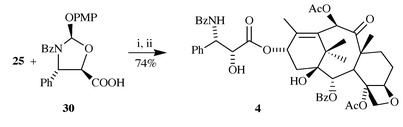
7-Deoxybaccatin is a direct precursor of paclitaxel analogues, as exemplified by its conversion into 7-deoxypaclitaxel (Scheme 6): esterification of 25 with acid 3016 (30 3 equiv., DCC 3 equiv., DMAP 0.5 equiv., THF, rt, 1 h), followed by acidic hydrolysis (5% p-TsOH in MeOH, rt, 1 h) afforded the title compound 49 in 74% yield.
We believe that the described chemistry offers an efficient pathway for the preparation of new paclitaxel derivatives, and points to the naturally abundant taxane diterpene fraction – taxine – as a valuable starting material for further semisynthetic studies.
Experimental
General remarks
All chromatographic separations were performed on Silica, 10–18, 60A, ICN Biomedicals. Standard techniques were used for the purification of reagents and solvents. NMR spectra were recorded on a Varian Gemini 200, 1H NMR at 200 MHz, 13C NMR at 50 MHz, for samples in deuterated chloroform. Chemical shifts are expressed in ppm using tetramethylsilane as internal standard, coupling constants (J) are in Hz. IR spectra were recorded on a Perkin-Elmer 457 grating FT instrument, and are expressed in cm−1. Optical rotations were measured on a Perkin-Elmer 141MC polarimeter; [α]D-values are given in units of 10−1 deg cm2 g−1. Mass spectra were obtained on a Finnigan ITDS 700 instrument. Microanalyses were performed at the Center for Instrumental Analysis, Faculty of Chemistry, University of Belgrade. Petroleum spirit refers to the fraction with distillation range 70–90 °C.Taxine 6 was obtained according to the previously reported procedure,7c in yields of 5–10 g kg−1 of dry leaves.
9-O-Acetyl-5-O-cinnamoyltaxicine I 8 and 10-O-acetyl-5-O-cinnamoyltaxicine 9
To a solution of taxine 6 (11.4 g) in THF (50 ml), was added methyl iodide (15 ml) dropwise, and the reaction mixture was stirred 4 h at rt. The solvent was removed under reduced pressure (T < 30 °C), the yellow powder (quaternary salt) was dissolved in THF (50 ml), and DBU (3 g) was added dropwise with stirring. The reaction mixture was stirred for 4 h, then the solvent was removed under reduced pressure (T < 30 °C), the mixture was partitioned between CH2Cl2 and water, the organic phase was washed successively with 1% HCl and water, and dried over anhydrous MgSO4. Evaporation of solvent gave 6–7 g of a yellow foam. Purification by flash chromatography (eluent: benzene–EtOAc 7∶3) afforded 1 g of 8 (less polar), 1.9 g of 9 (more polar), and 600 mg of a mixture of 8 + 9.1H and 13C NMR spectra for compounds 8 and 9 were identical to those previously reported.13
Isomerization of 8 into 9
A solution of 9-O-acetyl-5-O-cinnamoyltaxicine 8 (3.6 g) and KOAc (5 g) in methanol (50 ml) was stirred at rt for 12 h. The solvent was removed (Rotavap), the residue was partitioned between CH2Cl2 and water, and the organic extract was washed with water, dried over anhydrous MgSO4 and evaporated to dryness under reduced pressure. Purification as above afforded 700 mg of 8, 900 mg of 9, and 450 mg of a mixture of 8 + 9.10-O-Acetyl-5-O-cinnamoyltaxicine 1,2-carbonate 10
To a cold (0 °C) solution of phosgene (14.3 g) in CH2Cl2 (60 ml) was added dropwise a solution of 9 (1.95 g) and pyridine (2.85 g) in CH2Cl2 (20 ml) with stirring. After the reaction mixture had been stirred at 0 °C for 30 min, the solvent and excess of phosgene were removed under reduced pressure (T < 20 °C). The residue was dissolved in diethyl ether (100 ml), and water (60 ml) was added, followed by imidazole (50 mg). The reaction mixture was vigorously stirred 10 min at rt when TLC monitoring (SiO2 plates; developer benzene–EtOAc 7∶3) showed that hydrolysis of the chloroformate was complete. The organic layer was separated, the water layer was extracted with 3 × 50 ml CH2Cl2, and the combined organic extract was washed successively with dil. HCl and water, dried over anhydrous MgSO4 and evaporated to dryness under reduced pressure. Purification by dry-flash chromatography (eluent benzene–EtOAc 7∶3) afforded 1.8 g (90%) of 10 as a white foam, IR (KBr) νmax 3483, 3063, 2939, 1814, 1744, 1716, 1684, 1638, 1269, 1229, 1203, 1165, 1032, 770; 1H NMR δ 7.72 (m, 2H), 7.67 (d, J 16.3, 1H), 7.4 (m, 3H), 6.31 (d, J 16, 1H), 5.85 (d, J 9.5, 1H), 5.53 (s, 1H), 5.36 (superimposed 2 × s, 2H), 4.81 (d, J 5.75, 1H), 4.16 (d, J 9.5, 1H), 3.39 (d, J 5.62, 1H), 2.94 (s, 2H), 2.31 (s, 3H), 2.17 (s, 3H), 2.2–1.7 (m, 4H), 1.57 (s, 3H), 1.33 (s, 3H), 1.21 (s, 3H); 13C NMR δ 196.5 (C), 169.95 (C), 166.01 (C), 152.56 (C), 150.99 (C), 146.01 (CH), 141.71 (C), 139.93 (C), 134.36 (C), 130.47 (CH), 128.95 (2 × CH), 128.4 (2 × CH), 118.24 (CH2), 117.38 (CH), 88.76 (C), 79.43 (CH), 77.39 (CH), 76.37 (CH), 76.1 (CH), 44.98 (C), 43.15 (CH), 41.02 (C), 40.95 (CH2), 32.43 (CH3), 27.64 (CH2), 25.83 (CH2), 21.0 (CH3), 20.39 (CH3), 17.68 (CH3), 14.31 (CH3); MS/CIisobutane 565 (M + 1).9-O-Acetyl-5-O-cinnamoyltaxicine 1,2-carbonate 11
Compound 11 was obtained from 9-O-acetyl-5-O-cinnamoyltaxicine 8, using the same procedure as for the preparation of 10. Starting from 730 mg of 8, 860 mg of phosgene and 0.48 ml of pyridine in 17 ml of CH2Cl2, 609 mg (80%) of 11 was obtained. White foam, 1H NMR δ 7.73 (m, 2H), 7.65 (d, J 15.9, 1H), 7.43 (m, 3H), 6.31 (d, J 15.9, 1H), 5.63 (d, J 9.9, 1H), 5.54 (s, 1H), 5.37 (s, 1H), 5.34 (s, 1H), 5.07 (d, J 10.3, 1H), 4.95 (d, J 5.68, 1H), 3.39 (d, J 5.49, 1H), 2.96 (s, 2H), 2.19 (s, 3H), 2.16 (s, 3H), 2.04 (m, 2H), 1.76 (s, 3H), 1.7 (m, 2H), 1.42 (s, 3H), 1.01 (s, 3H); 13C NMR δ 196.83, 171.43, 166.03, 154.24, 152.58, 146.15, 139.67, 139.56, 134.35, 130.56, 129.0, 128.33, 118.72, 117.32, 88.94, 79.27, 78.47, 77.20, 71.73, 44.53, 43.06, 41.13, 32.84, 27.4, 27.06, 20.84, 19.96, 17.47, 14.40. Quaternary signal of C-8 could not be detected (probably superimposed with CH2 signal at δ 41.13).Isomerization of 11 into 10
A solution of carbonate 11 (6 g) and KOAc (10 g) in methanol (100 ml) was stirred 2 h at rt. Usual work-up, followed by flash chromatography (eluent benzene–EtOAc 4∶1) afforded 1.2 g of carbonate 10 (more polar), 1.32 g of recovered 11, and 0.83 g of a mixture of 10 + 11.Ketone 14 (Method A: Dess–Martin periodinane)
To a stirred suspension of periodinane17 (4.23 g) in CH2Cl2 (210 ml) was added a solution of 10 (1.6 g) in CH2Cl2 (50 ml), followed by a catalytic amount of trifluoroacetic acid (10 μl). The reaction mixture was stirred for 12 h at rt, then saturated aq. NaHSO3 (200 ml) was added, and stirring was continued for 45 min. The organic phase was washed successively with aq. NaHSO3 and brine, dried over anhydrous MgSO4, evaporated to dryness and purified by dry-flash chromatography (eluent: petroleum spirit–acetone 85∶15) to give ketone 14 (1.27 g, 80%) as a white foam, mp 192–193 °C (Calc. for C32H34O9: C, 68.31; H, 6.09. Found: C, 67.99; H, 5.84%); [α]20D +74.3 (c 1, CHCl3), IR (KBr) νmax 2928, 1821, 1752, 1713, 1687, 1638, 1229, 1204, 1164, 1020; 1H NMR δ 7.73 (m, 2H), 7.68 (d, J 16, 1H), 7.46 (m, 3H), 6.71 (s, 1H), 6.32 (d, J 16.1, 1H), 5.62 (d, J 1.4, 1H), 5.43 (br t, 1H), 5.40 (br s, 1H), 4.43 (d, J 5.5, 1H), 3.82 (d, J 5.5, 1H), 3.0 (s, 2H), 2.33 (s, 3H), 2.26 (s, 3H), 2.0 (m, 2H), 1.55 (m, 2H), 1.38 (s, 3H), 1.29 (s, 3H), 1.28 (s, 3H); 13C NMR δ 202.94 (C), 196.05 (C), 169.23 (C), 165.72 (C), 152.07 (C), 148.86 (C), 146.42 (CH), 141.85 (C), 138.72 (C), 134.15 (C), 130.62 (CH), 128.96 (2 × CH), 128.38 (2 × CH), 118.69 (CH2), 116.91 (CH), 88.6 (C), 80.99 (CH), 76.55 (CH), 76.19 (CH), 57.03 (C), 44.54 (CH), 41.31 (C), 40.51 (CH2), 31.54 (CH3), 31.01 (CH2), 27.33 (CH2), 20.67 (CH3), 18.32 (CH3), 15.28 (CH3), 14.75 (CH3), MS/CIisobutane 563 (M + 1).
Ketone 14 (Method B: PCC)
To a suspension of PCC (30 g) in CH2Cl2 (100 ml), was added a solution of 10 (2.9 g) in CH2Cl2 (30 ml), and the reaction mixture was vigorously stirred at rt overnight, then diluted with diethyl ether (200 ml), filtered through a pad of Florisil, and evaporated to dryness. Purification of the residue by dry-flash chromatography (eluent benzene–EtOAc 95∶5) afforded compound 14 (2.2 g, 75%), physical data as above.Ketone 14 (Method C: Jones reagent)
To a stirred, ice-cold (0 °C) solution of 10 (200 mg) in acetone (16 ml) was added Jones reagent18 (0.16 ml) dropwise. After the addition was complete the reaction mixture was stirred for 15 min at 0 °C then allowed to reach rt, and was stirred for an additional 30 min. Aq. NaHSO3 was added, the mixture was extracted with CH2Cl2, and the extract was dried over anhydrous MgSO4 and evaporated to dryness. Purification by dry-flash chromatography (eluent benzene–EtOAc 975∶25) afforded compound 14 (100 mg, 50%) physical data as above.
Tetraol 16
To a solution of ketone 14 (1.23 g) in a mixture of THF (23 ml) and water (11.5 ml), was added NMO (1.35 g), followed by osmium tetraoxide (1.85 ml of 2.5% solution in tBuOH). The reaction mixture was stirred 12 h at rt, then Florisil (0.6 g), sodium dithionite (120 mg) and water (9 ml) were added, and stirring was continued for 10 min. The reaction mixture was filtered, and the filtrate was thoroughly extracted with EtOAc. The organic extract was dried over anhydrous MgSO4, evaporated to dryness and purified by flash chromatography (eluent n-heptane–EtOAc 2∶3) to give compound 16 (1.37 g, quantitative) as a white foam, mp 158–159 °C; IR (KBr) νmax 3453, 2950, 2927, 2856, 1809, 1750, 1711, 1690, 1228, 1207, 1044, 1020; 1H NMR δ 7.55–7.3 (m, 5H), 6.59 (s, 1H), 5.14 (br s, 1H), 4.96 (d, J 2.95, 1H), 4.38 (2 × d superimposed, J1 4.5, J2 3.09, 2H), 3.98 (d, J 11.79, 1H), 3.89 (d, J 19.52, 1H), 3.58 (d, J 11.51, 1H), 3.27 (d, J 4.49, 1H), 2.85 (d, J 19.65, 1H), 2.26 (s, 3H), 2.24 (s, 3H), 2.1–1.8 (m, 4H), 1.33 (s, 3H), 1.27 (s, 3H), 1.22 (s, 3H); 13C NMR δ 202.86, 197.66, 172.86, 169.25, 152.90, 149.41, 142.34, 138.76, 128.32, 128.07, 126.63, 88.75, 81.75, 75.73, 75.72, 74.54, 74.10, 73.34, 62.74, 56.14, 45.23, 41.48, 40.46, 31.17, 30.0, 23.47, 20.65, 18.54, 16.64, 14.44; MS/CIisobutane 631 (M + 1).Triol 5 and (2S,3R)-(−)-(methyl 2,3-dihydroxy-3-phenylpropanoate) 17
To a solution of KOAc (0.75 g) in methanol (170 ml) was added tetraol 16 (1.16 g), and the mixture was heated to reflux with stirring for 22 min. The solvent was evaporated (Rotavap), the residue was diluted with water, extracted with CH2Cl2, and the extract was dried over anhydrous MgSO4 and evaporated to dryness under reduced pressure. Purification by dry-flash chromatography (eluent benzene–EtOAc 4∶1) afforded 230 mg of 17 (first eluted), and 665 mg (80%) of triol 5 (isolated as a monohydrate) as a white solid. Recrystallization of 17 from CH2Cl2–cyclohexane gave 190 mg (51%) of analytically pure 17, physical data identical with those previously reported.14Physical data for 5: white foam, mp >240 °C (decomp.) (Calc. for C23H30O10·H2O: C, 56.97; H, 6.60. Found: C, 57.28; H, 6.57%); [α]20D −108 (c 1, CHCl3), IR (KBr) νmax 3468, 2957, 1794, 1747, 1707, 1681, 1230, 1210, 1022; 1H NMR (in CDCl3 + D2O) δ 6.66 (s, 1H), 4.36 (d, J 4.63, 1H), 4.0 (d, J 10.5, 1H), 3.95 (d, J 19.37, 1H), 3.72 (br s, 1H), 3.57 (d, J 10.52, 1H), 3.52 (d, J 4.64, 1H), 2.84 (d, J 19.37, 1H), 2.23 (s, 3H), 2.22 (s, 3H), 2.2–1.6 (m, 3H), 1.35 (s, 3H), 1.25 (m, 1H), 1.22 (2 × s superimposed, 6H); 13C NMR δ 203.52 (C), 197.73 (C), 169.26 (C), 153.11 (C), 148.21 (C), 143.09 (C), 89.08 (C), 82.27 (CH), 75.74 (CH), 73.97 (CH), 70.0 (CH), 62.18 (CH2), 56.26 (C), 42.06 (CH), 41.58 (C), 40.52 (CH2), 31.1 (CH3), 28.99 (CH2), 24.16 (CH2), 20.7 (CH3), 18.57 (CH3), 16.67 (CH3), 14.28 (CH3), MS/CIisobutane 467 (M + 1).
Silyl derivative 19
A solution of triol 5 (83 mg), TBDMSCl (89 mg), triethylamine (65 mg) and DMAP (2.5 mg) in CH2Cl2 (2 ml) was stirred 24 h at rt under an argon atmosphere. The mixture was diluted with CH2Cl2, washed successively with ice-cold 1% HCl, aq. NaHCO3 and water, dried over anhydrous MgSO4, and the solvent was removed under reduced pressure. Purification by dry-flash chromatography (eluent benzene–EtOAc 9∶1) afforded silyl ether 19 (85.7 mg, 83%) as a colourless, viscous oil, and 9 mg of the starting triol 5 was recovered.Spectral data for 19: IR (film) νmax 3475, 3023, 2931, 1752, 1711, 1692, 1606, 1259, 1226, 1154, 1125, 1026; 1H NMR δ 6.66 (s, 1H), 4.31 (d, J 4.7, 1H), 3.98 (d, J 19.4, 1H), 3.97 (d, J 9.5, 1H), 3.96 (s, 1H, OH), 3.64 (br s, 1H), 3.53 (d, J 9.5, 1H), 3.52 (d, J 4.7, 1H), 2.83 (d, J 19.4, 1H), 2.78 (s, 1H, OH), 2.23 (s, 3H), 2.22 (s, 3H), 2.2–1.65 (m, 4H), 1.34 (s, 3H), 1.21 (s, 6H), 0.9 (s, 9H), 0.14 (s, 3H), 0.11 (s, 3H); 13C NMR δ 203.52 (C), 197.58 (C), 169.11 (C), 152.43 (C), 147.97 (C), 142.91 (C), 88.36 (C), 81.84 (CH), 75.65 (CH), 73.87 (C), 69.61 (CH), 62.98 (CH2), 56.12 (C), 41.52 (CH), 41.48 (C), 40.46 (CH2), 30.99 (CH3), 28.94 (CH2), 25.64 (CH3), 23.98 (CH2), 20.58 (CH3), 18.43 (CH3), 18.05 (C), 16.59 (CH3), 14.17 (CH3), −5.64 (CH3), MS/CIisobutane 581 (M + 1).
Mesylester 20
To an ice cold (0 °C), stirred solution of 19 (85.7 mg) in dry pyridine (1.85 ml) was added mesyl chloride (113 μl) dropwise. After the addition was complete, the cooling bath was removed, and the mixture was stirred 24 h at rt, diluted with CH2Cl2 (50 ml), washed successively with 2.25% HCl (11 ml), water, aq. NaHCO3 and water, and dried over anhydrous MgSO4. Evaporation under reduced pressure yielded crude ester 20 (120 mg, 97%), which was used in the next step without further purification.Mesylester 21
To a solution of 50% HF (0.35 g) in acetonitrile (7.8 ml) was added silyl derivative 20 (224.4 mg), and the reaction mixture was stirred for 6.5 h at rt, diluted with CHCl3, washed with aq. NaHCO3 and dried over anhydrous MgSO4. Purification by dry-flash chromatography (eluent benzene–EtOAc 3∶1) afforded mesylester 21 (149 mg, 80%) as a white powder, 1H NMR δ 6.66 (s, 1H), 4.84 (br s, 1H), 4.35 (d, J 4.4, 1H), 4.03 (d, J 11, 1H), 3.95 (d, J 19.3, 1H), 3.65 (d, J 10.9, 1H), 3.45 (d, J 4.4, 1H), 3.02 (s, 3H), 2.87 (d, J 19.4, 1H), 2.27 (s, 3H), 2.24 (s, 3H), 2.2–2.0 (m, 4H), 1.37 (s, 3H), 1.27 (s, 3H), 1.22 (s, 3H); 13C NMR δ 202.72 (C), 197.17 (C), 169.19 (C), 152.82 (C), 149.20 (C), 143.37 (C), 88.95 (C), 81.63 (CH), 81.46 (CH), 75.81 (CH), 73.03 (C), 62.0 (CH2), 56.03 (C), 43.43 (CH), 41.61 (C), 40.42 (CH2), 38.78 (CH3), 31.34 (CH3), 29.3 (CH2), 25.54 (CH2), 20.64 (CH3), 18.55 (CH3), 16.96 (CH3), 14.26 (CH3).Oxetane alcohol 22
A solution of mesyl compound 21 (149 mg) and diisopropylethylamine (247 mg) in toluene (40 ml) was heated to reflux with stirring for 39 h. The solvent was removed under reduced pressure, and the residue was submitted to dry-flash chromatography (eluent n-heptane–EtOAc 3∶2), to give compound 22 (100 mg, 82%) as a white solid, mp 128 °C; IR (KBr) νmax 3358, 2994, 2921, 1811, 1751, 1710, 1692, 1451, 1377, 1207, 1133, 1020, 960; 1H NMR δ 6.51 (s, 1H), 4.82 (dd, J1 8.65, J2 2.6, 1H), 4.66 (d, J 9.6, 1H), 4.52 (d, J 5.65, 1H), 4.48 (d, J 9.7, 1H), 3.71 (d, J 19.33, 1H), 2.84 (d, J 19.33, 1H), 2.76 (d, J 5.11, 1H), 2.25 (s, 3H), 2.11 (s, 3H), 2.15–1.9 (m, 1H), 1.72 (s, 3H), 1.7–1.6 (m, 2H), 1.35 (s, 3H), 1.26 (s, 3H); 13C NMR δ 203.66 (C), 197.27 (C), 169.26 (C), 152.83 (C), 148.92 (C), 142.4 (C), 88.36 (C), 87.18 (CH), 81.3 (CH), 79.69 (CH2), 75.97 (CH), 73.38 (C), 54.63 (C), 46.82 (CH), 41.42 (C), 40.27 (CH2), 31.97 (CH2), 30.88 (CH3), 26.56 (CH2), 20.59 (CH3), 18.56 (CH3), 14.27 (CH3), 14.23 (CH3), MS/CIisobutane 449 (M + 1).Acetate 23
A solution of the oxetane 22 (185 mg), DMAP (706 mg), and acetic anhydride (273 μl) in CH2Cl2 (50 ml) was stirred at rt for 4 h, then was diluted with CH2Cl2, washed successively with 1 M HCl, saturated aq. NaHCO3 and water, and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the residue was submitted to dry-flash chromatography (eluent benzene–EtOAc 9∶1), to give 126 mg (62.4%) of pure 23. Crystallization of impure fractions (20 mg) from CH2Cl2–n-heptane afforded an additional crop (7 mg, 3.5%) of the product. Total yield: 133 mg (66%).Compound 23 was obtained as a white solid, mp 275 °C (decomp.); [α]25D +17.1 (Calc. for C25H30O10: C, 61.22; H, 6.16. Found: C, 61.04; H, 6.00%); IR (KBr) νmax 3019, 2992, 2956, 1822, 1747, 1731, 1710, 1684, 1232, 1206, 1026, 772; 1H NMR δ 6.54 (s, 1H), 4.95 (d, J 8.74, 1H), 4.64 (d, J 8.91, 1H), 4.51 (d, J 5.46, 1H), 4.46 (dd, J1 9.1, J2 0.73, 1H), 3.47 (d, J 5.28, 1H), 2.96 (d, J 20, 1H), 2.84 (d, J 20, 1H), 2.25 (s, 3H), 2.1 (s, 3H), 2.08 (s, 3H), 2.08–1.95 (m, 2H), 1.77 (s, 3H), 1.75–1.65 (m, 2H), 1.34 (s, 3H), 1.25 (s, 3H); 13C NMR δ 203.41 (C), 195.58 (C), 170.38 (C), 169.1 (C), 152.09 (C), 149.98 (C), 142.89 (C), 88.23 (C), 83.27 (CH), 80.77 (CH), 79.9 (C), 76.1 (CH), 75.65 (CH2), 55.32 (C), 42.25 (CH), 41.37 (C), 39.8 (CH2), 31.92 (CH2), 31.25 (CH3), 27.0 (CH2), 21.55 (CH3), 20.67 (CH3), 18.52 (CH3), 14.47 (CH3), 14.12 (CH3), MS/CIisobutane 491 (M + 1).
Benzoate 24
Compound 24 was prepared from 23 according to the previously reported procedure.15 Starting from 126 mg of 23, after purification by dry-flash chromatography (eluent benzene–EtOAc 9∶1), 112 mg (77%) of benzoate 24 was obtained.Spectral data for compound 24 were identical with those previously reported.10d
7-Deoxybaccatin III 25
This was prepared from 24 according to the previously reported procedure.15 Starting from 18 mg of benzoate 24, after purification by dry-flash chromatography (eluent benzene–EtOAc 4∶1), compound 25 (12.5 mg, 69%; 83% based on consumed 24) was obtained; in addition, 3.1 mg of 24 was recovered.Physical data for compound 25 were identical with those previously reported.9b,c,10d
7-Deoxypaclitaxel 4
To a stirred solution of acid 3016 (137 mg) in THF (5 ml) was added DCC (71 mg). After 5 min a solution of 7-deoxybaccatin III 25 (65 mg) in THF (2 ml) was added, followed by DMAP (7 mg), and stirring was continued for 1 h, when TLC monitoring indicated completion of the reaction. The resulting suspension was filtered through a cotton plug, diluted with EtOAc, washed successively with aq. NaHCO3 and brine, dried over anhydrous MgSO4, and evaporated to dryness. The product was roughly purified by dry-flash chromatography (eluent benzene–EtOAc 85∶15), then was dissolved in methanol (10 ml), toluene-p-sulfonic acid (0.5 g) was added, and the mixture was stirred 2 h at rt, diluted with EtOAc, washed successively with aq. NaHCO3 and brine, dried over anhydrous MgSO4 and evaporated to dryness, and the residue was purified by dry-flash chromatography (eluent n-hexane–acetone 3∶1 → 3∶2). A second purification by flash chromatography (eluent CH2Cl2–methanol 95∶5) afforded 7-deoxypaclitaxel 4 (70 mg, 74% as a white powder, 1H NMR spectrum was identical with that previously reported.913C NMR spectrum of this compound has not been reported: δ 206.18 (C), 172.71 (C), 170.27 (C), 169.69 (C), 167.12 (C), 167.07 (C), 140.25 (C), 137.99 (C), 133.64 (CH), 133.60 (CH), 133.50 (C), 131.93 (CH), 130.22 (CH), 129.27 (C), 128.98 (CH), 128.71 (C), 128.65 (CH), 128.29 (CH), 127.05 (CH), 126.96 (CH), 84.50 (CH), 82.04 (C), 78.93 (C), 76.62 (CH2), 75.67 (CH), 74.07 (CH), 73.20 (CH), 72.19 (CH), 54.93 (CH), 52.75 (C), 44.99 (CH), 42.84 (C), 35.84 (CH2), 35.09 (CH2), 26.95 (CH2), 26.20 (CH3), 22.64 (CH3), 21.40 (CH3), 20.78 (CH3), 14.68 (CH3), 14.37 (CH3).
Diol 26 (interrupted osmylation of ketone 14)
The same method as that for 16 was used except that the reaction was interrupted after 1 h. Dry-flash chromatography (eluent benzene–EtOAc 9∶1 → 4∶1) afforded pure 26 and a mixture of 26 + 27 (27 could not be isolated in a pure state, but was tentatively detected in the mixture due to resonances at δ 7.75 and 6.4 (d, J 16) characteristic for the cinnamic double bond protons).Compound 26: white foam, 1H NMR δ 7.5–7.3 (m, 5H), 6.65 (s, 1H), 5.64 (d, J 1.41, 1H), 5.34 (s, 1H), 5.25 (br s, 1H), 4.88 (d, J 3.94, 1H), 4.41 (d, J 5.67, 1H), 4.22 (d, J 3.93, 1H), 3.53 (d, J 5.52, 1H), 2.98 (d, J 19.67, 1H), 2.87 (d, J 20.4, 1H), 2.26 (s, 3H), 2.25 (s, 3H), 2.12–1.85 (m, 2H), 1.6–1.4 (m, 2H), 1.36 (s, 3H), 1.26 (s, 3H), 1.24 (s, 3H); 13C NMR δ 202.77 (C), 196.76 (C), 171.89 (C), 169.25 (C), 151.99 (C), 148.95 (C), 141.92 (C), 138.81 (C), 137.83 (C), 128.32 (CH), 128.27 (CH), 126.39 (CH), 120.04 (CH2), 88.56 (C), 81.0 (CH), 78.97 (CH), 76.04 (CH), 75.18 (CH), 75.09 (CH), 56.85 (C), 44.88 (CH), 41.30 (C), 40.60 (CH2), 31.61 (CH3), 31.03 (CH2), 27.04 (CH2), 20.70 (CH3), 18.34 (CH3), 15.41 (CH3), 14.97 (CH3), MS/CIisobutane 597 (M + 1).
Ester 28 (isomerisation of tetraol 16 with KOAc)
To a solution of 16 (42.6 mg) in CH2Cl2 (3 ml) were added two drops of saturated aq. KOAc, followed by 18-crown-6 (1 mg), and the reaction mixture was vigorously stirred for 15 h at rt. Standard work-up afforded a quantitative yield of the C-20 ester 28 as a colourless film, 1H NMR (in CDCl3 + D2O) δ 7.4 (s, 5H), 6.65 (s, 1H), 4.97 (d, J 3.8, 1H), 4.64 (d, J 12, 1H), 4.40 (d, J 3.8, 1H), 4.35 (d, J 12, 1H), 4.3 (d, J 4.6, 1H), 3.8 (d, J 19.2, 1H), 3.75 (br s, 1H), 3.56 (d, J 4.6, 1H), 2.82 (d, J 19.2, 1H), 2.23 (s, 3H), 2.19 (s, 3H), 1.9–1.6 (m, 4H), 1.34 (s, 3H), 1.31 (s, 3H), 1.21 (s, 3H); 13C NMR δ 203.01 (C), 197.85 (C), 171.89 (C), 169.22 (C), 152.67 (C), 148.7 (C), 143.11 (C), 138.96 (C), 128.61 (CH), 128.3 (CH), 126.41 (CH), 88.95 (C), 81.99 (CH), 75.59 (CH), 75.15 (CH), 74.91 (CH), 74.82 (C), 68.65 (CH), 65.48 (CH2), 56.22 (C), 42.42 (CH), 41.56 (C), 40.44 (CH2), 31.2 (CH3), 28.86 (CH2), 24.4 (CH2), 20.67 (CH3), 18.56 (CH3), 16.63 (CH3), 14.21 (CH3), MS/CIisobutane 597 (M + 1).Acetate 29
To a solution of triol 5 (50 mg), pyridine (42 μl), and DMAP (1 mg) in CH2Cl2 (14 ml) was added acetic anhydride (13.3 μl), and the reaction mixture was stirred for 10 min at rt (longer reaction time results in the formation of the 5,20-diacetate). Standard work-up, followed by purification by dry-flash chromatography (eluent benzene–EtOAc 4∶1), afforded 20-acetate 29 (31.5 mg, 58%) as a colourless film, 1H NMR δ 6.68 (s, 1H), 4.52 (d, J 11.85, 1H), 4.42 (d, J 11.84, 1H), 4.37 (d, 4.89, 1H), 3.88 (d, J 19.42, 1H), 3.71 (br t, 1H), 3.32 (s, 1H, OH), 2.85 (d, J 18.8, 1H), 2.65 (br s, 1H, OH), 2.24 (s, 3H), 2.22 (s, 3H), 1.95–1.8 (m, 2H), 1.7–1.55 (m, 2H), 1.35 (s, 3H), 1.23 (s, 3H). For the results of the NOE experiment on C-20 acetate 29 see Fig. 1.Acknowledgements
R. N. S. is grateful to Professor Pierre Potier, Dr Alain Ahond, and Dr Christiane Poupat, from the Institut de Chimie des Substances Naturelles, Gif-sur-Yvette, for stimulating discussions.References
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325 CrossRef.
-
(a)
The Chemistry and Pharmacology of Taxol and its Derivatives, ed. V. Farina, Elsevier, Amsterdam, 1995;
Search PubMed;
(b)
TaxolR: Science and Applications, ed. M. Suffness, CRC Press, Boca Raton, 1995,
Search PubMedand references cited therein;;
(c) K. C. Nicolaou, W.-M. Dai and R. K. Guy, Angew. Chem., Int. Ed. Engl., 1994, 33, 15 CrossRef
Angew. Chem., 1994, 106, 38.
Search PubMed.
- (a) K. C. Nicolaou , H. Ueno , J.-J. Liu , P. G. Nantermet , Z. Yang , J. Renaud , K. Paulvannan and R. Chadha , J. Am. Chem. Soc., 1995, 117, 653, Search PubMedand references therein;; (b) R. A. Holton , H.-B. Kim , C. Somoza , F. Liang , R. J. Biediger , P. D. Boatman , M. Shindo , C. C. Smith , S. Kim , H. Nadizadeh , Y. Suzuki , C. Tao , P. Vu , S. Tang , P. Zhang , K. K. Murthi , L. N. Gentile and J. H. Liu , J. Am. Chem. Soc., 1994, 116, 1599, Search PubMedand references therein;; (c) S. J. Danishefsky, J. J. Masters, W. B. Young, J. T. Link, L. B. Snyder, T. V. Magee, D. K. Jung, R. C. A. Isaacs, W. G. Bornmann, C. A. Alaimo, C. A. Coburn and M. J. Di Grandi, J. Am. Chem. Soc., 1996, 118, 2843 CrossRef CAS; (d) P. A. Wender , N. F. Badham , S. P. Conway , P. E. Floreancig , T. E. Glass , J. B. Houze , N. E. Krauss , D. Lee , D. G. Marquess , P. I. McGrane , W. Meng , M. G. Natchus , A. J. Shuker , J. C. Sutton and R. E. Taylor , J. Am. Chem. Soc., 1997, 119, 2757, Search PubMedand references therein;; (e) T. Mukaiyama, I. Shiina, H. Iwadare, M. Saitoh, T. Nishimura, N. Ohkawa, H. Sakoh, K. Nishimura, Y. Tani, M. Hasegawa, K. Yamada and K. Saitoh, Chem. Eur. J., 1999, 5, 121 CrossRef CAS; (f) K. Morihara, R. Hara, S. Kawahara, T. Nishimori, N. Nakamura, H. Kusama and I. Kuwajima, J. Am. Chem. Soc., 1998, 120, 12980 CrossRef CAS.
- J.-N. Denis, A. E. Greene, D. Guenard, F. Gueritte-Voegelein, L. Mangatal and P. Potier, J. Am. Chem. Soc., 1988, 110, 5917 CrossRef CAS.
- D. Guenard, F. Gueritte-Voegelein and P. Potier, Acc. Chem. Res., 1993, 26, 160 CrossRef CAS.
- (a) G. Chauviere, D. Guenard, F. Picot, V. Senilh and P. Potier, C. R. Seances Acad. Sci., Ser. 2, 1981, 293, 501 Search PubMed; (b) K. M. Witherup, S. A. Look, M. W. Stasko, T. J. Ghiorzi, G. M. Mushik and G. M. Cragg, J. Nat. Prod., 1990, 53, 1249 CAS; (c) ref. 4. .
- (a) H. Lucas, Arch. Pharm., 1856, 14, 438 Search PubMed; (b) G. Appendino , in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon, 1996, vol. 11, p. 237; Search PubMed; (c) L. H. D. Jenniskens, E. L. M. van Rosendaal, T. A. van Beek, P. H. G. Wiegerinck and H. W. Scheeren, J. Nat. Prod., 1996, 59, 117 CrossRef CAS; (d) for an alternative isolation procedure see ref. 10a..
- (a) L. Ettouati, A. Ahond, C. Poupat and P. Potier, J. Nat. Prod., 1991, 54, 1455 CrossRef CAS; (b) In some specimens of Taxus baccata the content of taxine is as high as 17 g kg−1, 10 g being taxine B; we are grateful to Dr A. Ahond and Dr C. Poupat for this personal communication. .
- (a) A. G. Chaudhary, J. M. Rimoldi and D. G. I. Kingston, J. Org. Chem., 1993, 58, 3798 CrossRef CAS; (b) S.-H. Chen, S. Huang, J. Kant, C. Fairchild, J. Wei and V. Farina, J. Org. Chem., 1993, 58, 5028 CrossRef CAS; (c) V. Farina , S.-H. Chen , D. R. Langley , M. D. Wittman , J. Kant and D. Vyas , Eur. Pat. Appl. EP 590 267, 6 Apr. 1994, (Chem. Abstr., 1994, 121, 205747n). Search PubMed.
- (a) L. Ettouati, A. Ahond, C. Poupat and P. Potier, Tetrahedron, 1991, 47, 9823 CrossRef CAS; (b) P. H. G. Wiegerinck, L. Fluks, J. B. Hammink, S. J. E. Mulders, F. M. H. de Groot, H. L. M. van Rosendaal and H. W. Sheeren, J. Org. Chem., 1996, 61, 7092 CrossRef CAS; (c) I. Fenoglio, G. M. Nano, D. G. V. Velde and G. Appendino, Tetrahedron Lett., 1996, 37, 3203 CrossRef CAS; (d) H. Poujol , A. A. Mourabit , A. Ahond , C. Poupat and P. Potier , Tetrahedron, 1997, 53, 12575, Search PubMedand references therein;.
- While this work was in progress, the first semisynthesis of 7-deoxybaccatin from 3 was reported, in 15 steps, and 1.7% overall yield (ref. 10d)..
- R. Matović and R. N. Saičić, Chem. Commun., 1998, 1745 RSC.
- G. Appendino, P. Gariboldi, A. Pisetta, E. Bombardelli and B. Gabetta, Phytochemistry, 1992, 31, 4253 CrossRef CAS.
- J.-N. Denis, A. Correa and A. E. Greene, J. Org. Chem., 1990, 55, 1957 CrossRef CAS.
- K. C. Nicolaou, P. G. Nantermet, H. Ueno, R. K. Guy, A. Couladouros and E. J. Sorensen, J. Am. Chem. Soc., 1995, 117, 624 CrossRef CAS.
- A. M. Kanazawa, J.-N. Denis and A. E. Greene, J. Chem. Soc., Chem. Commun., 1994, 2591 RSC.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- A. Bowers, T. G. Halsall, E. R. H. Jones and A. J. Lemin, J. Chem. Soc., 1953, 2548 RSC.
- N. F. Magri, D. G. I. Kingston, C. Jitrangsri and T. Piccariello, J. Org. Chem., 1986, 51, 3239 CrossRef CAS.
Footnotes |
| † In this reaction yields varied between 60 and 94%. It is interesting to compare these reaction conditions with those needed for the hydrolysis of structurally related cinnamic esters, which require 20 M NaOH at reflux, for many days.10a,b For a study on selective hydrolysis of the paclitaxel side-chain, with the retention of C-10 acetate, see ref. 19. |
| ‡ Other reagents tried include: TBAF–THF, KF–18-crown-6–THF, KOAc–MeOH, NaH–THF, DBU and pyridine. Attempts to effect the cyclization via the corresponding triflate3c gave a complex mixture from which the oxetane was isolated in 4% yield. |
| This journal is © The Royal Society of Chemistry 2000 |