Bi–KOH. An efficient reagent for the coupling of nitroarenes to azo and azoxy compounds
Dhrubojyoti Dey
Laskar
,
Dipak
Prajapati
and
Jagir S.
Sandhu
*
Regional Research Laboratory, Jorhat, 785006, Assam, India
First published on 24th December 1999
Abstract
A simple and inexpensive procedure for the coupling of nitroarenes to azoxy compounds with bismuth and potassium hydroxide in methanol at ambient temperature is achieved. When carried out under microwave irradiation it gives exclusively azo compounds in excellent yields.
Application of bismuth metal to organic synthesis has recently attracted much attention due to its potential as a reductant for various synthetic purposes.1 Among group Va elements, bismuth is inexpensive, easy to handle and less toxic than arsenic and antimony and can be expected to play a crucial role in organic synthesis owing to its metallic character. However, to our knowledge, the use of bismuth in synthesis has scarcely been studied.2 Herein we wish to disclose the first example of Bi–KOH used for the reductive coupling of nitroarenes to azo and azoxy arenes in high yields. There are many methods for the preparation of azoxy compounds by the reduction of nitro compounds,3 but side reactions (e.g. dehalogenation, polymerisation) usually accompany the reductions, and so their use is limited.
The use of bismuth in organic synthesis is of particular interest as there are some naturally occurring azoxybenzenes which possess potent biological properties.4 Also, metal catalysed reactions are of interest because of their relevance to the enzymatic degradation of nitrogen-containing compounds in biological synthesis.5 Recently we have reported the rapid pinacolization of aromatic carbonyl compounds with bismuth and KOH in methanol at ambient temperature.6 To explore its synthetic utility we have investigated the reductive coupling of nitro compounds with bismuth and KOH in methanol at room temperature and under microwave irradiation (Scheme 1).
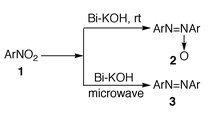 | ||
| Scheme 1 | ||
Results and discussion
In a typical case, nitrobenzene, bismuth powder and methanol were placed in a round-bottomed flask and stirred at ambient temperature and solid KOH was added. The progress of the reaction was monitored by TLC. After completion, followed by usual work-up of the mixture, p,p′-dichloroazoxybenzene was produced in 90% yield, and there was no evidence for the formation of N-phenylhydroxylamine or p,p′-dichloroazobenzene or p-chloroaniline. Various substituted nitrobenzenes were then reduced to the corresponding azoxy compounds; typical results are shown in Table 1. Nitrobenzene did not react with bismuth in methanol in the absence of KOH or when KOH was replaced by NH4Cl, indicating the significance of the basicity of the reaction. The replacement of KOH by NaOH, however, led to only a very minor reaction with the recovery of most of the nitrobenzene. The use of other metals such as magnesium or cadmium in combination with potassium hydroxide are also not effective and they produce a mixture of products along with totally reduced primary amines. In the case of methyl 4-nitrocinnamate the reaction occurred selectively at the nitro group and the carbon–carbon double bond in the substrate was not affected. High yields of azoxyarenes can be obtained from nitroaromatics with ether or alkyl substituents, the positional relationship of the substituent in no way influencing the overall reaction. Both bromo- and iodo-substituted nitro aromatics reacted smoothly with retention of the halogen, instead of its elimination.7 The carbonyl group in nitroacetophenone remained intact and the corresponding p,p′-diacetylazoxybenzene was obtained in 80% yield exclusively.8| Entry | Ar![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) a a |
Time/h (at rt) | Yieldb (%) |
Mp/°C | Time/min (mw) | Yield (%) |
|---|---|---|---|---|---|---|
| a Products were identified by the comparison of IR and NMR spectra and melting points with those of authentic samples. b Yield refers to the yield of pure isolated products. c Increasing the time of reaction had no significant effect on the yield and resulted in a small amount of decomposition. | ||||||
| 1 | Ph | 6 | 90 | 35–3614a |
8 | 85 |
| 2 | 4-ClC6H4 | 7 | 80 | 154–15614a |
8 | 82 |
| 3 | 2-ClC6H4 | 8 | 82 | 53.5–5514a |
8 | 70 |
| 4 | 4-BrC6H4 | 6 | 85 | 173–17514c |
7 | 85 |
| 5 | 3-ClC6H4 | 8 | 80 | 96–9714a |
8 | 78 |
| 6 | 4-MeC6H4 | 7 | 85 | 66–6814a |
7 | 80 |
| 7 | 4-EtC6H4 | 8 | 80 | 110–11214b |
8 | 80 |
| 8 | 4-OMeC6H4 | 8 | 80 | 116–11814c |
7 | 78 |
| 9 | 4-IC6H4 | 7 | 78 | 209–21014d |
6 | 82 |
| 10 | 4-MeCOC6H4 | 6 | 80 | 192–19414g |
8 | 83 |
| 11 | 4-MeOOCCH![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CHC6H4 CHC6H4 |
8 | 82 | 57–5814f |
7 | 75 |
| 12 |
|
8 | 75 | 138–13914e |
8 | 60e |
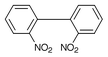
It is also remarkable to note that when the above reaction was carried out under thermal conditions, i.e. refluxed for 45 min, it yields only azoxy compound 2 (Table 1, entry 1) in 60% yield. Further increasing the reaction time upto 8 h produces a mixture of azo compound 3 in 75% yield along with azoxy compound in about 10% yield. Increasing the reaction time had no significant effect on the yield, rather decomposition occurs. We have also carried out the same reaction under microwave irradiation and observed the formation of azo compound 3 (entry 1) exclusively in 85% yield and there was no evidence for the formation of any azoxy compounds. Interestingly, it was also observed that this reduction system can be conveniently used to perform reductive intramolecular cyclisation of o,o′-dinitrobiphenyl to the corresponding benzo[c]cinnoline N-oxide 5 in good yield at room temperature (Scheme 2). When the reaction mixture was heated for 50 min it gave mixtures of benzo[c]cinnoline 6 and it’s N-oxide 5. Therefore the reaction is general (Table 1) and can be employed for the synthesis of symmetrically substituted chloro, bromo, iodo, alkoxy and alkyl azoxy compounds. All the compounds obtained were identified by comparision of IR and NMR spectra and melting points with those of authentic samples.
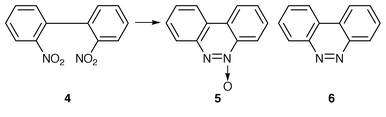 | ||
| Scheme 2 | ||
In conclusion, the present new method employing the metallic bismuth and KOH system has some notable advantages compared with other methods. For example in the reduction of bromo- or iodo-nitrobenzene by Mg, Tl or Al reagents, the halogens were usually eliminated or complex products were formed9 and in the reduction of alkyl substituted nitrobenzene by sodium alcoholate, polymers were usually formed.10 In contrast, in our examples none of these side reactions were observed. Therefore the present method because of its simplicity and high selectivity, constitutes a useful alternative to the commonly accepted procedure for the synthesis of various azo- and azoxybenzenes11 and benzo[c]cinnoline N-oxides.12
Experimental
Mps were measured in open capillary tubes on a Buchi melting point apparatus and are uncorrected. IR spectra were recorded as KBr discs on a Perkin-Elmer 240C analyser. The 1H NMR spectra were recorded with a Varian T-60 machine using tetramethylsilane (TMS) as the internal standard. The chemical shifts are recorded as δ values. Bismuth powder was of commercial grade and obtained from Loba Chemie Indoaustralian Co., Bombay. All other chemicals were purified by distillation or crystallisation prior to use.Conversion of nitroarenes to azoxyarenes at ambient temperature: general procedure
Nitrobenzene (1.23 g, 10 mmol), bismuth powder (4.18 g, 20 mmol) and methanol (6 ml) were placed in a 50 ml round-bottomed flask mounted over a magnetic stirrer. The reaction mixture was stirred at ambient temperature and KOH (3.37 g, 60 mmol) was added. The progress of the reaction was monitored by TLC. After 8 h of stirring, the reaction was quenched by adding diethyl ether (30 ml) to the reaction flask. The contents of the flask were stirred and the diethyl ether solution was decanted. The process was repeated twice with diethyl ether (2 × 15 ml) to extract the product from the methanol layer. The combined ether extract (60 ml) was washed with 10% HCl (3 × 15 ml), dried over anhydrous sodium sulfate and the solvent was distilled off to give almost the pure product. Further purification was achieved by column chromatography by using hexane–toluene (3∶2) as eluent to give azoxybenzene (yellow crystals) in 90% yield. Similarly other substituted nitroarenes were reacted and their characteristics are recorded in the table.Conversion of nitroarenes to azoxyarenes under thermal condition
Nitrobenzene (1.23 g, 10 mmol), bismuth powder (4.18 g, 20 mmol) and methanol (6 ml) were placed in a 50 ml round-bottomed flask fitted with a reflux condenser. KOH (3.36 g) was added to the contents and the resulting mixture was refluxed (at 60–65 °C) for 45 min. The reaction mixture was then cooled and quenched by adding 30 ml of diethyl ether. The contents were stirred and the diethyl ether solution was decanted off. The process was repeated twice with diethyl ether (2 × 15 ml). The combined ether extract was washed with 10% HCl (3 × 20 ml) and dried over anhydrous sodium sulfate. The solvent was distilled and the obtained product was purified by chromatography using hexane–toluene (3∶2) as eluent to give the pure azoxybenzene in 60% yield.Conversion of nitroarenes to azoarenes under thermal conditions
Nitrobenzene (1.23 g, 10 mmol), bismuth powder (4.18 g, 20 mmol) and methanol (6 ml) were placed in a 50 ml round- bottomed flask, KOH (3.37 g) was added to the flask and the resulting mixture was refluxed for 6–8 h. The reaction mixture was then cooled and quenched by adding 30 ml of diethyl ether. The contents were stirred and the diethyl ether solution was decanted off. The process was repeated twice by adding diethyl ether (2 × 15 ml). The combined ether extract was washed with 10% HCl and dried over anhydrous sodium sulfate. The solvent was distilled off and the products obtained were separated by chromatography using hexane–toluene (4∶1) as eluent to give azobenzene13 in 75% yield and azoxybenzene in 10% yield.Conversion of nitroarenes to azoarenes using Bi–KOH under microwave irradiation: general procedure
Nitrobenzene (1.23 g), bismuth powder (2.08 g, 10 mmol), KOH (1.68 g, 30 mmol) and 5 ml of MeOH were taken in an Eyrlenmeyer flask and placed in a commercial microwave oven (operating at 2450 MHz frequency) and irradiated for 6–8 min. The reaction mixture was allowed to cool and 30 ml of diethyl ether was added; the ethereal solution was decanted off. The process was repeated twice and the combined ethereal extract was washed with 10% HCl and dried over anhydrous sodium sulfate. The solvent was distilled off to give the almost pure azo compound in 85% yield, mp 68–69 °C. Further purification was achieved by chromatography using benzene–petroleum ether as eluent. Similarly other substituted nitroarenes were reacted and their characteristics are recorded in Table 1.References
- (a) M. Wada, H. Ohki and K.-Y. Akiba, J. Chem. Soc., Chem. Commun., 1987, 708 RSCand references cited therein; (b) H. Tanaka, A. Kosaka, S. Yamashita, K. Morisaki and S. Torii, Tetrahedron Lett., 1989, 1261 CrossRef CAS; (c) M. Wada, E. Takeichi and T. Matsumoto, Bull. Chem. Soc. Jpn., 1991, 64, 990 CASand references cited therein..
- Recently, Barton reported the functional group oxidation and phenylation by pentavalent organobismuth reagents: (a) D. H. R. Barton, J. C. Blazejewski, B. Charpiot, J. P. Finet, W. B. Motherwell, M. T. B. Papoula and S. P. Stanforth, J. Chem. Soc., Perkin Trans. 1, 1985, 2667 RSC; (b) D. H. R. Barton and J. P. Finet, Pure Appl. Chem., 1987, 59, 937 CAS.
- For some notable examples of azoxy compounds formation see: (a) A. McKillop and R. A. Raphad, J. Org. Chem., 1970, 35, 1671; (b) A. Osuka, H. Shimizu and H. Suzuki, Chem. Lett., 1983, 1373 CAS; (c) A. Bassant, M. Prato, P. Rampazzo, U. Quintily and G. Scorrano, J. Org. Chem., 1980, 45, 2263 CrossRef; (d) J. P. Sayder, V. T. Bandurco, F. Darack and H. Olsen, J. Am. Chem. Soc., 1974, 96, 5158 CrossRef; (e) U. Quintily and G. Scorrano, J. Org. Chem., 1983, 48, 3022 CrossRef.
- S. Murata, M. Miura and N. Nomura, J. Org. Chem., 1989, 54, 4700 CrossRef.
- J. P. Snyder, V. T. Bandurco, F. Durack and H. Olsen, J. Am. Chem. Soc., 1974, 96, 5158 CrossRef.
- B. Baruah, D. Prajapati and J. S. Sandhu, Tetrahedron Lett., 1995, 36, 6747 CrossRef CAS.
- A. McKillop and R. A. Raphad, J. Org. Chem., 1970, 35, 1671.
- For the reduction of the carbonyl group to hydroxy in azoxybenzene formation see: K. Oke, H. Takahashi, S. Uemura and N. Sugita, J. Chem. Soc., Chem. Commun., 1988, 591 RSC.
- (a) K. F. Katrstead, Can. J. Chem., 1933, 31, 1064; (b) J. F. Corbett, Chem. Commun., 1968, 1257 Search PubMed.
- (a) I. Shimao, Nippon Kagaku Kaishi., 1974, 515 Search PubMed; (b) C. M. Sutter and F. B. Danis, J. Am. Chem. Soc., 1928, 50, 2733 CrossRef.
- For pre-existing methods see: K. F. Keirstead, Can. J. Chem., 1953, 31, 1064 CAS; P. T. Lansbury and J. O. Peterson, J. Am. Chem. Soc., 1963, 85, 2236 CrossRef CAS; H. C. Brown, P. M. Welssman and N. M. Yoon, J. Am. Chem. Soc., 1966, 66, 1458 CrossRef; R. O. Hutchins, D. W. Lamson, L. Rue, C. Milewski and B. Maryanoff, J. Org. Chem., 1971, 36, 803 CrossRef CAS; T. S. Suzuki and T. Okada, Chem. Ind., 1970, 1926 Search PubMed.
- For some recent reports see: H. R. Sonawane, A. V. Pol, P. P. Moghe, S. S. Biswas and A. Sudalai, J. Chem. Soc., Chem. Commun., 1994, 1215 RSC; S. Sakaue, T. Tsubakino, Y. Nishiyama and Y. Ishii, J. Org. Chem., 1993, 58, 3633 CrossRef CAS; C. Paradisi, G. Gonzalez-Trueba and G. Scorrano, Tetrahedron Lett., 1993, 34, 877 CrossRef CAS.
- A. Alberti, N. Bedogni, A. H. Benaglia, R. Leardini, N. Nahui, G. J. Pedulli, A. Tiendo and G. Zanardi, J. Org. Chem., 1992, 57, 607 CrossRef CAS.
- For literature melting points see: (a) P. H. Gore and O. H. Weelar, J. Am. Chem. Soc., 1956, 78, 2160 CrossRef CAS; (b) E. Bamberger, Chem. Ber., 1926, 59, 481 Search PubMed; (c) R. S. Porter and J. E. Johnson, J. Phys. Chem., 1962, 66, 1826 CAS; (d) E. Bamberger and W. Hama, Justus Liebigs, Ann. Chem., 1873, 165, 189 Search PubMed; (e) F. Ullman and P. Dielere, Ber. Dtsch. Chem. Ges., 1904, 37, 23 Search PubMed; (f) D. Vorlander, Ber., 1906, 39, 803 Search PubMed; (g) H. Tani, S. Tanaka and F. Toda, Bull. Chem. Soc. Jpn., 1963, 36, 1267 CAS.
| This journal is © The Royal Society of Chemistry 2000 |