Diastereoselective addition reactions of racemic chiral vinyl sulfimides
Charlotte P.
Rayner (née Baird)
,
Andrew J.
Clark
,
Stuart M.
Rooke
,
Tim J.
Sparey
and
Paul C.
Taylor
*
Department of Chemistry, University of Warwick, Coventry, UK CV4 7AL
First published on 12th January 2000
Abstract
S-Aryl S-vinyl sulfimides are prepared by a new method via Horner–Wadsworth–Emmons reagents generated in situ. Lewis acid-catalysed Diels–Alder reaction of S-ethenyl-S-phenyl-N-tosylsulfimide with cyclopentadiene leads to the endo anti product with good diastereofacial and endo–exo selectivities. A range of alcohols and amines, with the limitation that they be practically usable in large excess, add to β-aryl vinyl sulfimides with modest diastereoselectivity. The major products of both the cycloaddition and the nucleophilic additions are rationalised by a model invoking addition to the least-hindered face of the s-cisconformation of the vinyl sulfimide.
Introduction
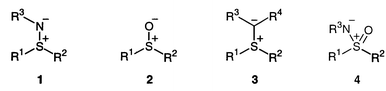
Compounds 1, which have the IUPAC name “sulfimide” and the Chemical Abstractsname “sulfilimine”, remain little-studied in the area of stereoselective synthesis,1 as compared with, for example, sulfoxides 2,2 sulfur ylides 3![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 3 and sulfoximides (sulfoximines) 4.4 However, there have been a number of recent reports regarding the synthesis of enantiomerically enriched sulfimides1,5 and their application in enantioselective synthesis of non-racemic epoxides and aziridines,6 which represent a small renaissance of interest in this field.
3 and sulfoximides (sulfoximines) 4.4 However, there have been a number of recent reports regarding the synthesis of enantiomerically enriched sulfimides1,5 and their application in enantioselective synthesis of non-racemic epoxides and aziridines,6 which represent a small renaissance of interest in this field.
The asymmetric chemistry of chiral vinyl sulfoxides and sulfoximides is now well-established,2,4 but the parallel chemistry of sulfimides remains largely unexplored. We decided to study the following classes of addition to the S-vinyl group of a sulfimide:
(i) Diels–Alder additions of dienes;
(ii) additions of nucleophiles;
(iii) additions of carbon-centred radicals.
To our knowledge, previous to our work there were no examples of classes (i) and (iii) in the sulfimide series. However, there are a number of reports from Yamamoto and co-workers of additions of alcohols, thiols, amines and doubly-stabilised carbanions to β-unsubstituted sulfimides,7 which are useful precedents for our work on the second class (nucleophilic additions). In the β-unsubstituted examples studied by the Yamamoto group, no new chiral centre is established. We envisaged carrying out analogous reactions on β-substituted vinyl sulfimides to establish the stereochemical influence of the sulfimidyl group.
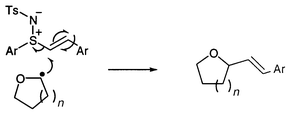
Our initial work on the third class of reactions (radical additions) has already been communicated.8 In this case, the major product appears to result from α-addition with subsequent elimination of the sulfimidyl group (Scheme 1). A comparative study of reactions of radicals with various vinyl sulfur compounds has recently been completed and will be published separately.9
 | ||
| Scheme 1 | ||
Herein we describe (i) a short study on the Diels–Alder reactions of two aryl vinyl sulfimides, (ii) the results of nucleophilic additions to a series of new β-substituted aryl vinyl sulfimides and (iii) a stereochemical model for the addition reactions.
Results and discussion
(i) Diels–Alder additions
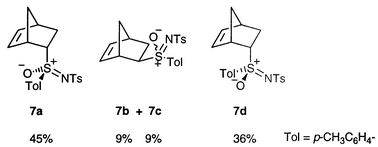
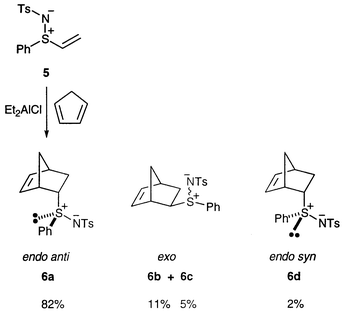
Lewis acid-catalysed reaction of 5 with cyclopentadiene was attempted in toluene at room temperature, using some of the Lewis acids proposed for the analogous cycloadditions of substituted vinyl sulfoxides.10 With TiCl4 a complex mixture of unidentified products resulted, but with ZnCl2 a mixture of cycloadducts was isolated in low yield, in which an endo product appeared to be dominant.Finally, using Et2AlCl at room temperature, a 55% isolated yield of a mixture of the four possible stereoisomers 6, which were not separated, was achieved (Scheme 2). The 1H NMR spectrum of the purified mixture was compared with those of the analogous sulfoxides11 and sulfoximides,12 which resulted from uncatalysed reactions at relatively high temperature. Not surprisingly, the closest fit, as judged by comparison of chemical shifts, was with the sulfoximides 7 (which differ only in the oxo group at sulfur and a para methyl group on the non-sulfonyl S-aryl group) and we thus used comparison with 7 to tentatively assign structures to the four isomers.
 | ||
| Scheme 2 | ||
The chemical shift data for the major isomer 6a are a reasonable match for those of both of the endo sulfoximides 7a and 7d (Table 1) and we thus confidently assign the endo stereochemistry to 6a. Closer inspection reveals an excellent match between the data for 6a and sulfoximide 7a, with the greatest discrepancies being with those protons (H-1 and H-3endo) that would be expected to be the most affected by the oxo group. The configuration of another closely analogous sulfoximide was established by the Glass group by X-ray crystallography12 and we thus believe the assignment of the relative stereochemistry of the sulfur centre to be secure. This leads us to speculate that the major product in the sulfimide case is the endo anti compound 6a (adapting the terminology proposed by Montanari and co-workers13,14 and referring to approach past the S–N rather than S–O bond).
|
|
|||
|---|---|---|---|
| 6a δ/ | 7a δ/ | 7d δ/ | |
| H-1 | 2.44 | 2.69 | 3.57 |
| H-2 | 3.78 | 3.96 | 3.93 |
| H-3 endo | 1.32 | 1.69 | 1.33 |
| H-3 exo | 2.13 | 2.23 | 1.78 |
| H-4 | 2.93 | 2.97 | 2.92 |
| H-5 & H-6 | 5.97, 6.29 | 5.79, 6.21 | 5.82, 6.14 |
| H-7 syn | 1.25 | 1.25 | 1.27 |
| H-7 anti | 1.49 | 1.48 | 1.49 |
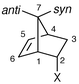
The data for the 11% and 5% isomers cannot be extracted completely from the spectrum, making unambiguous structural assignments difficult. Nevertheless it can be observed that the data for these two compounds are (a) quite similar and (b) best matched to the two exo sulfoximide analogues 7b and 7c. Hence we tentatively assign the exo stereochemistry to isomers 6b and 6c. Assignment of the relative stereochemistry of the sulfur centre is not possible from these data; isolation and either crystallography or stereospecific oxidation to the sulfoximide would be required. Finally, by a process of elimination we assign the endo syn stereochemistry to the minor isomer 6d, but there are no independent data to confirm this.
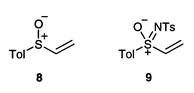
If our assignments are correct, the Lewis acid-catalysed reaction of vinyl sulfimide 5 with cyclopentadiene proceeds with significant endo anti selectivity. Comparison with the thermal reactions of the corresponding sulfoxide 8 and sulfoximide 9 shows that the endo selectivity is as expected (Table 2). However, there is a significant change in both the magnitude and sense of the diastereofacial selectivity (Table 2). We have followed De Lucchi in extending the syn/anti nomenclature to sulfoximides by reference to the oxo group rather than the imido group,14 but note that this is necessarily somewhat artificial. The variations in syn/anti selectivity (if correctly assigned) need not be surprising considering the differences in reaction conditions (115 °C for the sulfoxide, refluxing dichloromethane for the sulfoximide and Et2AlCl at room temperature for the sulfimide). Indeed, prior conversion of the vinyl sulfoxide to a sulfoxonium salt leads to the endo anti product with good stereoselectivities.11c
| endo∶exo | syn∶anti | |
|---|---|---|
| a Assuming 11% exo isomer is syn, otherwise 13∶87. | ||
| Sulfoxide 8 | 64∶36 | 50∶50 |
| Sulfoximide 9 | 81∶19 | 55∶45 |
| Sulfimide 5 | 84∶16 | 7∶93a |
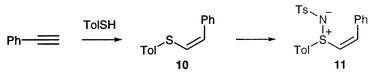
We proceeded to attempt the same Et2AlCl-catalysed reaction of the Z-β-substituted analogue 11. Sulfimide 11 was prepared by imidation of the known sulfide 10 which is the isomerically pure product of addition of thiocresol to phenylacetylene (Scheme 3). Reaction with cyclopentadiene under our previous best conditions led to a crude product with signals in the 1H NMR spectrum at around 6.0 and 6.45 ppm (cf. 6.0 and 6.3 ppm for the alkene protons in 11) and no signal for the upfield vinylic proton of the starting material 11 at 6.3 ppm. However, on work-up only unreacted starting material 11 could be isolated. It seems that the Diels–Alder reaction does indeed occur, but that a retro-reaction occurs during work-up to give back the starting material. This observation is significant since, as pointed out by Paquette and Magnus,15 low yields in sulfoxide-mediated Diels–Alder reactions are often attributed to low reactivity, without consideration of the possibility of the existence of a relatively fast equilibrium. It may be that in our case the Et2AlCl is not a true catalyst (three equivalents were used), but remains coordinated to the product, thus preventing the reverse reaction occurring until the Lewis acid is removed.
 | ||
| Scheme 3 | ||
We wished to extend the cycloaddition chemistry to acyclic dienes, but starting material was recovered from reactions of both sulfimides 5 and 11 with Danishefsky’s diene 12.
(ii) Nucleophilic additions to vinyl sulfimides
As mentioned in the introduction, we were interested in the stereochemical influence of the sulfimidyl group on nucleophilic additions to the β-vinylic carbon of sulfimides of general structure 13. Since the sulfimidyl group is readily reduced to the corresponding sulfide,16 additions of alcohols and amines would lead to derivatives of β-hydroxy- and β-aminosulfides respectively, which have potential as chiral ligands for metals.17 Hence, it was important that our chemistry was readily adaptable to the enantiomerically pure analogues.The two main literature routes to vinyl sulfimides are direct imidation of sulfides, as used in Section (i) above, of which no asymmetric version yet exists for vinyl analogues,5a and elimination from β-halo sulfimides.18 Although a good ee was reported for one example of an asymmetric version of this elimination reaction (Scheme 4),18b we did not feel that this procedure had general potential.
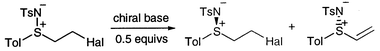 | ||
| Scheme 4 | ||
At the start of our work, one of the few readily-available enantiomerically-pure sulfimides was compound 14 prepared from the corresponding commercially available enantiomerically-pure sulfoxide using the procedure of Cram et al.19 We therefore decided to use this methyl sulfimide as our starting material and to adapt the method used by Craig’s group for the synthesis of related vinyl sulfoximides 15.20 This involves the in situ generation of Horner–Wadsworth–Emmons type reagents from the methyl sulfoximide and their reaction with aldehydes (Scheme 5).
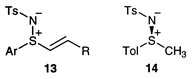
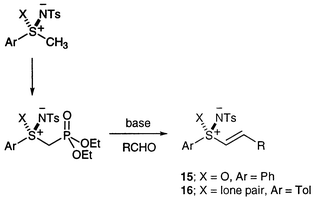 | ||
| Scheme 5 | ||
Using the identical conditions to Craig’s group,20 we were able to isolate a range of new racemic chiral β-substituted aryl vinyl sulfimides 16 (Table 3), albeit in lower yield than observed with the sulfoximides. In all cases the (E) isomer was the major product, but the selectivities varied considerably with the substituent. The aryl derivatives gave separable (E)∶(Z) mixtures, in which the predominance of the (E) product increased with increasing electron demand of the substituent (Table 3), whereas for the isopropyl and tert-butyl compounds, the (Z) isomer was not observed. However, another alkyl derivative, the cyclopropyl product 16g, showed virtually no selectivity.
Although all the products described here are racemic we did undertake a small-scale preparation of 16b using enantiomerically-enriched 14 and determined that the ees of product and starting material were the same (by integration of the p-tosyl methyl signals in the 1H NMR spectrum in the presence of trifluoroanthrylethanol). Hence this method should be applicable to asymmetric synthesis.
The reactions of some of the new aryl sulfimides with some representative alcohols and amines are summarised in Scheme 6 and Table 4. Unfortunately, the effect of the β-substituent meant that these reactions only gave useful yields, as compared with the unsubstituted literature examples,7 when the alcohol or amine was used as the solvent; a similar phenomenon was reported by Pyne et al. in the sulfoxide series.21 Hence, the scope of this reaction is limited to cheap, volatile alcohols and amines which can feasibly be used as solvents.
| X | Y | R | Yield (%) | 17∶18 | |
|---|---|---|---|---|---|
| a Isolated yield of 17. b Mixture of 17 and 18. c Major isomer not identified. | |||||
| a | O | H | PhCH2 | 64a |
>97∶3 |
| b | O | H | Et | 73a |
90∶10 |
| c | O | OMe | Et | 32b |
90∶10 |
| d | NH | H | PhCH2 | 63a |
80∶20 |
| e | NH | Cl | PhCH2 | 57a |
80∶20 |
| f | NH | H | i-Pr | 53b |
80∶20 |
| g | NH | Cl | i-Pr | 69b |
80∶20 |
| h | NEt | H | Et | 46b |
70% dec |
| i | NEt | Cl | Et | 56b |
70% dec |
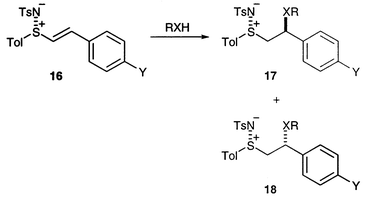 | ||
| Scheme 6 | ||
The diastereoselectivities of the reactions were modest (Table 4), with the exception of the addition of benzyl alcohol which proceeded in at least 95% de. The stereochemistry of the major isomer was inferred by comparison with some sulfoxide analogues 19.22 The benzylic proton in Pyne et al.’s examples of (R,S) 19 are doublets of doublets with coupling constants of around 3.0 and 10.5 Hz, in excellent agreement with our major isopropylamine adducts 17f and 17g (3.0 and 10.5 Hz for both examples) and the major benzylamine adducts 17d and 17e (2.3 and 10.7 Hz approximately). Furthermore, the major alcohol adducts 17a–c have even more disparate coupling constants (2.1 and 10.9 Hz approximately). We therefore propose the (RS,SR) stereochemistry for all these adducts.
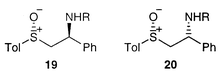
The same coupling constants for the diastereoisomeric (R,R) sulfoxides 20 are less easy to extract from the literature data, but can be seen to be significantly closer to each other in value. With sulfimides 18f and 18g we were able to interpret the 1H NMR data for the minor isomer; the coupling constants for the benzylic proton (dd, J 7.2, 7.2 Hz in both cases) resembled those of sulfoxides 20 and thus gave support to our stereochemical assignments. The equivalent multiplets for the diethylamine adducts 17h, 17i, 18h and 18i have a different pattern, presumably due to the significant increase in the steric demand of the amino group, and we are thus reluctant to assign the relative stereochemistry of these adducts.
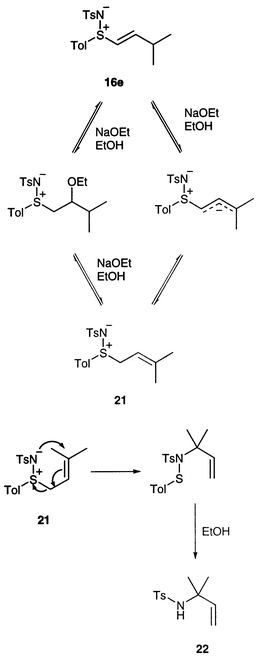
Additions of alcohols and amines to the tert-butyl derivative 16f were unsuccessful, presumably due to steric hindrance. However, the isopropyl derivative 16e did react with ethanol, but with a different result. In this case the product was the known sulfonamide 22 which we presume to have been formed via alkene 21 (Scheme 7). Rearrangements of closely related allylic sulfimides are well-known.5a,23
 | ||
| Scheme 7 | ||
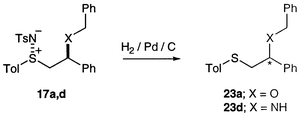
Finally, we wished to demonstrate that the chiral sulfimide products 17 could be readily transformed to sulfides, to permit synthesis of potentially useful chiral ligands. The most challenging substrates were the benzyl alcohol and benzylamine adducts 17a, 17d and 17d, since selective hydrogenolysis of the S–N bond in the presence of the benzyl groups was required. In fact, with both 17a and 17d the reduction proceeded cleanly to furnish the target sulfides 23 with the O- and N-benzyl groups intact (Scheme 8).
 | ||
| Scheme 8 | ||
(iii) Rationalisation of the sense of stereoselectivity
Pyne and co-workers have remarked that one simple model can account for the majority of stereoselective reactions of vinyl sulfoxides.24 Direct extension of this model leads to Fig. 1, where the reactive conformation of the vinyl sulfimide is s-cis and addition occurs from the least hindered face. In the cycloaddition, there is a need to invoke the usual endo preference and it should be noted that a Lewis acid is also involved in this case.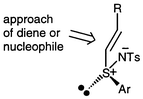 | ||
| Fig. 1 | ||
The model correctly predicts (i) the anti cycloadduct 6a to be the major Diels–Alder product and (ii) the observed outcome of the addition of amines and alcohols to yield the (RS,SR) major products 17 (as with vinyl sulfoxides21).
Conclusion
S-Aryl S-vinyl sulfimides were prepared by a literature route and by a new method starting from S-aryl S-methyl sulfimides via Horner–Wadsworth–Emmons reagents generated in situ, this route being appropriate for the synthesis of enantiomerically enriched vinyl sulfimides.Lewis acid-catalysed Diels–Alder reaction of one unsubstituted vinyl sulfimide with cyclopentadiene was successful, leading to the endo anti product with good diastereofacial and endo–exo selectivities, but extension to other examples failed. A range of alcohols and amines, with the limitation that they be practically usable in large excess, add to β-aryl vinyl sulfimides with modest diastereoselectivity. The major products of both the cycloaddition and the nucleophilic additions are predicted by a model invoking addition to the least-hindered face of the s-cis conformation of the vinyl sulfimide.
Experimental
General
Melting points were determined on a Stuart Scientific SMP1 melting point apparatus and are uncorrected. Microanalyses were performed at the University of Warwick. Mass spectra were recorded on a Kratos MS90 spectrometer. Infra-red spectra were recorded on a Perkin-Elmer 1720X Fourier transform spectrometer. NMR spectra were recorded on Bruker ACF 250 or Bruker ACP 400 instruments. Flash chromatography was performed on silica gel (Merck Kieselgel 60F254, 230–400 mesh). Racemic methyl sulfimide 14 was prepared by the literature route.23a
S-Ethenyl-S-phenyl-N-p-tosylsulfimide 5![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) 18a
18a
Phenyl vinyl sulfide (1.0 g, 7.3 mmol) and glacial acetic acid (0.2 cm3, 3.5 mmol) were added to a solution of chloramine-T hydrate (2.07 g, 7.3 mmol) in absolute ethanol (200 cm3). The mixture was stirred at 40 °C for 1 hour. The mixture was cooled to room temperature and stirred overnight. The solvent was removed and water (100 cm3) and dichloromethane (100 cm3) were added. The organic layer was separated, dried over anhydrous magnesium sulfate and concentrated in vacuo to give the crude product. Trituration with ether followed by filtration and flash chromatography on silica gel using chloroform–ethyl acetate (8∶1) as eluant furnished white crystals of sulfimide 5 (1.01 g, 45%), mp 109–110 °C (lit.,18a 111–113 °C); Rf 0.61 (EtOAc) (Found: C, 59.02; H, 4.94; N, 4.55. C15H15NO2S2 requires C, 59.02; H, 4.92; N, 4.59%); νmax(CHCl3)/cm−1 1599, 1494, 1478, 1446, 1298, 1284, 1143, 1089, 962; δH(250 MHz; CDCl3) 2.34 (3H, s, Me), 6.03 (1H, dd, J = 8.4, 0.6 Hz, H-trans), 6.29 (1H, dd, J = 15.6, 0.6 Hz, H-cis), 6.41 (1H, dd, J = 8.4, 15.6 Hz, CHS), 7.17 (2H, AA′ of AA′BB′, J = 8.5 Hz, tosyl), 7.44–7.55 (3H, m, meta and para Ph), 7.64 (2H, m, ortho Ph), 7.75 (2H, BB′ of AA′BB′, J = 8.5 Hz, tosyl); δC(62.9 MHz; CDCl3) 21.26 (Me), 126.02 (2), 126.80 (2), 129.10 (2), 129.68, 129.90 (2), 132.47, 132.78, 134.36, 141.18, 141.67; m/z (CI; NH3) 306 (65%, MH+), 198 (38), 189 (100), 155 (24), 137 (100), 108 (64), 91 (61), 65 (21).
Reaction of ethenyl phenyl sulfimide 5 with cyclopentadiene
Diethylaluminium chloride (0.3 cm3 of a 1 M solution in hexanes, 0.30 mmol) was added to a solution of freshly distilled cyclopentadiene (0.10 cm3, 0.76 mmol) and vinyl sulfimide 5 (30 mg, 0.10 mmol) in dry toluene (10 cm3) under nitrogen at 0 °C. The mixture was stirred at 0 °C for 2 hours and then at room temperature for a further 12 hours. The solvent was removed under vacuum and the residue was subjected to flash chromatography on silica gel using chloroform–ethyl acetate (4∶1) as eluant furnishing white crystals of S-norbornenyl-S-phenyl-N-tosylsulfimide 6 in a ratio of 82∶11∶5∶2 (endo anti∶exo∶exo∶ endo syn), (20 mg, 55%); Rf (CHCl3): 0.32 (Found: C, 64.41; H, 5.52; N, 3.44. C20H21NO2S2 requires C, 64.69; H, 5.66; N, 3.77%).![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH), 6.29 (1H, dd, J = 5.7, 3.1 Hz, CH), 7.04 (2H, AA′ of AA′BB′, J = 8.1 Hz, tosyl), 7.46 (2H, m, Ph), 7.50 (1H, m, Ph), 7.59 (2H, BB′ of AA′BB′, J = 8.2 Hz, tosyl), 7.68 (2H, m, Ph); δC(100.6 MHz; CDCl3) 21.38 (Me), 29.69 (C-3), 42.30 (C-1/C-4), 44.53 (C-1/C-4), 49.38 (C-7), 66.93 (C-2), 126.23 (2), 127.37 (2), 128.96 (2), 129.90 (2), 131.43, 132.71, 135.36, 139.72, 141.33, 141.44.
CH), 6.29 (1H, dd, J = 5.7, 3.1 Hz, CH), 7.04 (2H, AA′ of AA′BB′, J = 8.1 Hz, tosyl), 7.46 (2H, m, Ph), 7.50 (1H, m, Ph), 7.59 (2H, BB′ of AA′BB′, J = 8.2 Hz, tosyl), 7.68 (2H, m, Ph); δC(100.6 MHz; CDCl3) 21.38 (Me), 29.69 (C-3), 42.30 (C-1/C-4), 44.53 (C-1/C-4), 49.38 (C-7), 66.93 (C-2), 126.23 (2), 127.37 (2), 128.96 (2), 129.90 (2), 131.43, 132.71, 135.36, 139.72, 141.33, 141.44.
(Z)-Phenylethenyl p-tolyl sulfide 1025
To a solution of sodium (1.85 g, 80.4 mmol) in absolute ethanol (100 cm3) was added thiocresol (10.0 g, 80.5 mmol) and phenylacetylene (9.70 cm3, 80.4 mmol). The mixture was refluxed for 2 days. After standing for a further 2 days, the mixture was poured into water (100 cm3) and dichloromethane (100 cm3) was added. The organic layer was separated, and the aqueous layer was extracted twice with dichloromethane (100 cm3). The combined organic layers were dried over anhydrous magnesium sulfate and concentrated in vacuo, giving exclusively sulfide 10 (14.72 g, 81%), mp 62–64.5 °C (lit.,25 64–65.5 °C); Rf (MeOH): 0.80 (Found: C, 79.57; H, 6.22. C15H14S requires C, 79.64; H, 6.19%); νmax(CHCl3)/cm−1 1599, 1588, 1494, 1445, 1358, 1094, 1019, 849, 810; δH(250 MHz; CDCl3) 2.42 (3H, s, Me), 6.56 (1H, d, J = 10.8 Hz, CHPh), 6.64 (1H, d, J = 10.8 Hz, CHS), 7.23 (2H, AA′ of AA′BB′, J = 7.9 Hz, p-tolyl), 7.30 (5H, m, Ph), 7.65 (2H, BB′ of AA′BB′, J = 7.9 Hz, p-tolyl); δC(62.9 MHz; CDCl3) 21.10 (Me), 126.49 (2), 127.02 (2), 127.05 (2), 128.32 (2), 128.74, 129.96, 130.50, 132.67, 136.58, 137.38; m/z (EI) 226 (100%, M+), 211 (30), 91 (25), 77 (23).
(Z)-S-2-Phenylethenyl-S-p-tolyl-N-p-tosylsulfimide 11
) and (Z) sulfimides (10∶90) were obtained which on trituration yielded (Z)-11 (7.95 g, 75%); Rf (CHCl3): 0.35; mp 123.5–125 °C (Found: C, 66.32; H, 5.33; N, 3.61. C22H21NO2S2 requires C, 66.84; H, 5.32; N, 3.54%); νmax(CHCl3)/cm−1 1597, 1492, 1446, 1297, 1284, 1143, 1088, 967; δH(250 MHz; CDCl3) 2.32 (3H, s, tolyl Me), 2.38 (3H, s, tosyl Me), 6.33 (1H, d, J = 10.0 Hz, CHPh), 7.10 (2H, AA′ of AA′BB′, J = 7.8 Hz, tolyl), 7.13 (1H, d, J = 10.0 Hz, CHS), 7.27 (2H, AA′ of AA′BB′, J = 7.8 Hz, tosyl), 7.38 (5H, m, Ph), 7.54 (2H, BB′ of AA′BB′, J = 7.8 Hz, tolyl), 7.70 (2H, BB′ of AA′BB′, J = 7.8 Hz, tosyl); δC(62.9 MHz; CDCl3) 21.30 (2 × Me), 126.05 (2), 126.24 (2), 128.76 (2), 128.99 (2), 129.43 (2), 130.09, 130.51 (2), 132.42, 132.58, 141.20, 141.36, 141.40, 142.77 (one C missing); m/z (CI; NH3) 396 (5.4%, MH+), 227 (100), 226 (74), 189 (34), 124 (28), 108 (21), 91 (45).
(E)-S-[2-(4-Methoxyphenyl)ethenyl]-S-p-tolyl-N-p-tosylsulfimide 16a
To a solution of S-methyl-S-p-tolyl-N-p-tosylsulfimide 14 (1.0 g, 3.26 mmol) in THF (100 cm3) at −78 °C under nitrogen were added dropwise n-BuLi (2.5 M, 1.3 cm3, 3.26 mmol), potassium tert-butoxide (1.0 M, 3.27 cm3, 3.26 mmol) and diethyl chlorophosphate (0.47 cm3, 3.26 mmol). After stirring for 10 min, p-anisaldehyde (0.4 cm3, 3.26 mmol) in THF (2 cm3) was added dropwise and the reaction left to warm to 0 °C over 30 min. Saturated aqueous ammonium chloride (100 cm3) was added, along with sufficient water to dissolve any salts precipitated, and the reaction extracted with dichloromethane (3 × 80 cm3). The combined organic layers were washed with water (150 cm3) and dried over magnesium sulfate, filtered and concentrated under reduced pressure. A 1H NMR spectrum of the crude product (60% yield) showed an (E∶Z) ratio of 80∶20. Column chromatography of the yellow oil on silica using ether as eluant separated the stereoisomers. The major product was the (E) isomer 16a (0.67 g, 48%), mp 122–124 °C; Rf 0.30 (ether) (Found: C, 64.91; H, 5.45; N, 3.29. C23H23NO3S2 requires C, 64.81; H, 5.39; N, 3.23%); νmax(Nujol)/cm−1 1602, 1512, 1465, 1307, 1293, 1170, 1136, 1087, 959, 802; δH(250 MHz; CDCl3) 2.29 (3H, s, Me tolyl), 2.36 (3H, s, Me tosyl), 3.79 (3H, s, MeO), 6.42 (1H, dd, J = 15.1, 0.9 Hz, CHAr), 6.85 (2H, AA′ of AA′BB′, MeOC6H4), 7.13 (AA′ of AA′BB′, tolyl), 7.25 (AA′ of AA′BB′, tosyl), 7.26 (1H, d, J = 15.1 Hz, CHS), 7.30 (2H, BB′ of AA′BB′, MeOC6H4), 7.55 (BB′ of AA′BB′, tolyl), 7.76 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 21.3 (2 × Me), 55.4 (MeO), 114.3, 119.8 (CHAr), 125.3, 126.2, 126.6, 129.0, 129.7, 130.5, 132.3, 141.3, 141.4, 141.5, 142.9, 161.6 (CHS); m/z (EI) 426 (2%, MH+), 256 (100), 241 (21), 211 (35), 91 (76).
(E)-S-(2-Phenylethenyl)-S-p-tolyl-N-p-tosylsulfimide 16b
To a solution of S-methyl-S-p-tolyl-N-p-tosylsulfimide 14 (4.0 g, 0.013 mol) in THF (300 cm3) at −78 °C under nitrogen were added dropwise n-BuLi (2.5 M, 5.2 cm3, 0.013 mol), potassium tert-butoxide (1.0 M, 13 cm3, 0.013 mol) and diethyl chlorophosphate (1.88 cm3, 2.0 mol). After stirring for 10 min, benzaldehyde (1.32 cm3, 0.013 mmol) in THF (5 cm3) was added dropwise and the reaction left to warm to 0 °C over 30 min. Saturated aqueous ammonium chloride (300 cm3) was added, along with sufficient water to dissolve any salts precipitated, and the reaction extracted with dichloromethane (3 × 150 cm3). The combined organic layers were washed with water (300 cm3) and dried over magnesium sulfate, filtered and concentrated under reduced pressure. A 1H NMR spectrum of the crude product (48% yield) showed an (E∶Z) ratio of 85∶15. Column chromatography of the yellow oil on silica using chloroform–ethyl acetate 4∶1 as eluant separated the stereoisomers. The major product was the (E) isomer 16b (1.95 g, 38%), mp 121–122 °C; Rf 0.33 (ether) (Found: C, 66.80; H, 5.35; N, 3.54. C22H21NO2S2 requires C, 66.90; H, 5.33; N, 3.46%); νmax(Nujol)/cm−1 1662, 1601, 1491, 1307, 1138, 661; δH(400 MHz; CDCl3) 2.30 (3H, s, Me tolyl), 2.37 (3H, s, Me tosyl), 6.55 (1H, d, J = 15.2 Hz, CHPh), 7.14 (2H, AA′ of AA′BB′, tolyl), 7.26 (2H, AA′ of AA′BB′, tosyl), 7.32 (1H, d, J = 15.2 Hz, CHS), 7.35 (5H, m, Ph), 7.55 (2H, BB′ of AA′BB′, tolyl), 7.76 (2H, BB′ of AA′BB′, tosyl); δC(100.6 MHz; CDCl3) 21.30 (Me tolyl), 21.38 (Me tosyl), 122.70 (CHPh), 126.23, 126.82, 128.91, 129.07, 129.43, 130.56, 130.76, 132.00, 132.70, 141.07 (CHS), 141.40, 141.54, 143.20; m/z (FAB) 396 (100%, MH+), 227 (92), 226 (100).
The minor isomer (Z)-S-(2-phenylethenyl)-S-p-tolyl-N-p-tosylsulfimide 11 was also isolated (9%), mp 124–125 °C; spectra identical to 11 above.
(E)-S-[2-(4-Chlorophenyl)ethenyl]-S-p-tolyl-N-p-tosylsulfimide 16c
To a solution of S-methyl-S-p-tolyl-N-p-tosylsulfimide 14 (4.0 g, 0.013 mol) in THF (300 cm3) at −78 °C under nitrogen were added dropwise n-BuLi (2.5 M, 5.2 cm3, 0.013 mol), potassium tert-butoxide (1.0 M, 13 cm3, 0.013 mol) and diethyl chlorophosphate (1.88 cm3, 2.0 mol). After stirring for 10 min, benzaldehyde (1.32 cm3, 0.013 mmol) in THF (5 cm3) was added dropwise and the reaction left to warm to 0 °C over 30 min. Saturated aqueous ammonium chloride (300 cm3) was added, along with sufficient water to dissolve any salts precipitated, and the reaction extracted with dichloromethane (3 × 150 cm3). The combined organic layers were washed with water (300 cm3) and dried over magnesium sulfate, filtered and concentrated under reduced pressure. A 1H NMR spectrum of the crude product (40% yield) showed an (E∶Z) ratio of 95∶5. Column chromatography of the yellow oil on silica using chloroform–ethyl acetate 4∶1 as eluant was used to isolate the (E) isomer (1.95 g, 38%), mp 108–109 °C; Rf 0.43 (ether) (Found: C, 61.16; H, 4.66; N, 3.18. C22H20ClNO2S2 requires C, 61.45; H, 4.69; N, 3.18%); νmax(Nujol)/cm−1 1652, 1491, 1307, 1138, 955, 803, 774, 661; δH(400 MHz; CDCl3) 2.32 (3H, s, Me tolyl), 2.40 (3H, s, Me tosyl), 6.54 (1H, d, J = 15.1 Hz, CHPh), 7.16 (2H, AA′ of AA′BB′, tolyl), 7.26 (1H, d, J = 15.1 Hz, CHS), 7.31 (6H, m, tosyl and p-ClC6H4), 7.55 (2H, BB′ of AA′BB′, tolyl), 7.76 (2H, BB′ of AA′BB′, tosyl); δC(100.6 MHz; CDCl3) 21.4 (Me tolyl and tosyl), 123.26 (CHPh), 126.11, 126.80, 129.02, 129.07, 129.21, 131.22, 131.61, 132.07, 136.32, 139.07, 141.36 (CHS), 141.58, 143.21; m/z (FAB) 430 (49%, MH+).
(E)-S-(3-Methylbut-1-enyl)-S-p-tolyl-N-p-tosylsulfimide 16e
To a solution of S-methyl-S-p-tolyl-N-p-tosylsulfimide 14 (1.0 g, 3.26 mmol) in THF (100 cm3) at −78 °C under nitrogen were added dropwise n-BuLi (2.5 M, 1.3 cm3, 3.26 mmol), potassium tert-butoxide (1.0 M, 3.27 cm3, 3.26 mmol) and diethyl chlorophosphate (0.47 cm3, 3.26 mmol). After stirring for 10 min, isobutyraldehyde (0.30 cm3, 3.26 mmol) in THF (1 cm3) was added dropwise and the reaction left to warm to 0 °C over 30 min. Saturated aqueous ammonium chloride (100 cm3) was added, along with sufficient water to dissolve any salts precipitated, and the reaction extracted with dichloromethane (3 × 80 cm3). The combined organic layers were washed with water (150 cm3) and dried over magnesium sulfate, filtered and concentrated under reduced pressure. A 1H NMR spectrum of the crude product (64% yield) showed an (E∶Z) ratio of >98∶2. Column chromatography of the yellow oil on silica using ether as eluant separated the stereoisomers. The major product was the (E) isomer 16e (0.41 g, 35%), mp 108–110 °C; Rf 0.37 (ether); νmax(Nujol)/cm−1 1602, 1490, 1284, 1138, 1088, 1021, 962; δH(250 MHz; CDCl3) 0.97 (6H, 2 × d, J = 7.0 Hz, Me), 2.33 (3H, s, Me tolyl), 2.36 (3H, s, Me tosyl), 2.41 (1H, m, CHMe), 5.94 (1H, dd, J = 15.0, 1.3 Hz, CHS), 6.55 (1H, dd, J = 15.0, 6.4 Hz, CHR), 7.15 (AA′ of AA′BB′, tolyl), 7.25 (AA′ of AA′BB′, tosyl), 7.47 (BB′ of AA′BB′, tolyl), 7.72 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 20.8 (Me), 20.9 (Me), 21.3 (2 × ArMe), 31.3 (CHMe), 123.1 (CHR), 126.2, 126.5, 129.0, 130.4, 132.0, 141.1, 141.5, 142.9, 151.7 (CHS); m/z (CI/NH3) 362 (100%, MH+).
(E)-S-(3,3-Dimethylbut-1-enyl)-S-p-tolyl-N-p-tosylsulfimide 16f
To a solution of S-methyl-S-p-tolyl-N-p-tosylsulfimide 14 (1.0 g, 3.26 mmol) in THF (100 cm3) at −78 °C under nitrogen were added dropwise n-BuLi (2.5 M, 1.3 cm3, 3.26 mmol), potassium tert-butoxide (1.0 M, 3.27 cm3, 3.26 mmol) and diethyl chlorophosphate (0.47 cm3, 3.26 mmol). After stirring for 10 min, pivalaldehyde (0.354 cm3, 3.26 mmol) in THF (1 cm3) was added dropwise and the reaction left to warm to 0 °C over 30 min. Saturated aqueous ammonium chloride (100 cm3) was added, along with sufficient water to dissolve any salts precipitated, and the reaction extracted with dichloromethane (3 × 80 cm3). The combined organic layers were washed with water (150 cm3) and dried over magnesium sulfate, filtered and concentrated under reduced pressure. A 1H NMR spectrum of the crude product (39% yield) showed an (E∶Z) ratio of >98∶2. After column chromatography of the yellow oil on silica using ether as eluant, the major product was the (E) isomer 16f (0.48 g, 39%), mp 146–147 °C; Rf 0.46 (ether) (Found: C, 63.96; H, 6.76; N, 3.73. C20H25NO2S2 requires C, 63.82; H, 6.66; N, 3.71%); νmax(Nujol)/cm−1 1598, 1492, 1284, 1141, 1089, 1021, 962; δH(250 MHz; CDCl3) 0.98 (9H, s, Me), 2.32 (3H, s, Me tolyl), 2.36 (3H, s, Me tosyl), 5.89 (1H, d, J = 15.0 Hz, CHS), 6.53 (1H, d, J = 15.0 Hz, CHR), 7.15 (AA′ of AA′BB′, tolyl), 7.25 (AA′ of AA′BB′, tosyl), 7.47 (BB′ of AA′BB′, tolyl), 7.73 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 21.3 (2 × ArMe), 28.3 (Me), 126.2 (CHR), 126.5, 126.6, 129.0, 130.4, 132.1, 141.4, 141.6, 142.9, 155.3 (CHS); m/z (CI/NH3) 376 (100%, MH+), 220 (43), 207 (26), 206 (17).
S-(2-Cyclopropylethenyl)-S-p-tolyl-N-p-tosylsulfimide 16g
To a solution of S-methyl-S-p-tolyl-N-p-tosylsulfimide 14 (614 mg, 2.0 mmol) in THF (40 cm3) at −78 °C under nitrogen were added dropwise n-BuLi (2.5 M, 0.80 cm3, 2.0 mmol), potassium tert-butoxide (1.0 M, 2.0 cm3, 2.0 mmol) and diethyl chlorophosphate (0.29 cm3, 2.0 mmol). After stirring for 10 min, cyclopropanecarbaldehyde (0.149 cm3, 2.0 mmol) in THF (1 cm3) was added dropwise and the reaction left to warm to 0 °C over 30 min. Saturated aqueous ammonium chloride (40 cm3) was added, along with sufficient water to dissolve any salts precipitated, and the reaction extracted with dichloromethane (3 × 50 cm3). The combined organic layers were washed with water (100 cm3) and dried over magnesium sulfate, filtered and concentrated under reduced pressure. A 1H NMR spectrum of the crude product (39% yield) showed an (E∶Z) ratio of 53∶47. Column chromatography of the yellow oil on silica using ether as eluant led to a mixture of inseparable stereoisomers (0.28 g, 39%) (Found: MH+ 360.1104. C20H25NO2S2 requires 360.1092); νmax(Nujol)/cm−1 1606, 1292, 1280, 1142, 1089, 981, 734, 661.![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )-S-(2-Cyclopropylethenyl)-S-p-tolyl-N-p-tosylsulfimide 16g..
δ
H(250 MHz; CDCl3) 0.59 (2H, m, CH2), 0.99 (2H, m, CH2), 2.03 (1H, m, CH), 2.36 (3H, s, Me tolyl), 2.39 (3H, s, Me tosyl), 5.58 (1H, dd, J = 10.8, 9.0 Hz, CHR), 6.04 (1H, dd, J = 9.0, 0.6 Hz, CHS), 7.18 (AA′ of AA′BB′, tolyl), 7.28 (AA′ of AA′BB′, tosyl), 7.57 (BB′ of AA′BB′, tolyl), 7.79 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 8.7 (CH2), 9.1 (CH2), 12.4 (CH), 21.3 (2 × ArMe), 123.9 (CHR), 125.9, 126.3, 129.0, 130.4, 132.6, 141.3, 141.7, 142.3, 150.6 (CHS).
)-S-(2-Cyclopropylethenyl)-S-p-tolyl-N-p-tosylsulfimide..
δ
H(250 MHz; CDCl3) 0.58 (2H, m, CH2), 0.93 (2H, m, CH2), 1.53 (1H, m, CH), 2.35 (3H, s, Me tolyl), 2.38 (3H, s, Me tosyl), 6.01 (1H, dd, J = 10.8, 14.5 Hz, CHR), 6.02 (1H, d, J = 14.5 Hz, CHS), 7.16 (AA′ of AA′BB′, tolyl), 7.26 (AA′ of AA′BB′, tosyl), 7.49 (BB′ of AA′BB′, tolyl), 7.74 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 8.7 (2 × CH2), 14.5 (CH), 21.3 (2 × ArMe), 121.2 (CHR), 126.2, 126.5, 129.0, 130.4, 132.3, 141.3, 141.6, 142.7, 150.7 (CHS); m/z (FAB) 360 (100%, MH+), 190 (14), 123 (46).
)-S-(2-Cyclopropylethenyl)-S-p-tolyl-N-p-tosylsulfimide 16g..
δ
H(250 MHz; CDCl3) 0.59 (2H, m, CH2), 0.99 (2H, m, CH2), 2.03 (1H, m, CH), 2.36 (3H, s, Me tolyl), 2.39 (3H, s, Me tosyl), 5.58 (1H, dd, J = 10.8, 9.0 Hz, CHR), 6.04 (1H, dd, J = 9.0, 0.6 Hz, CHS), 7.18 (AA′ of AA′BB′, tolyl), 7.28 (AA′ of AA′BB′, tosyl), 7.57 (BB′ of AA′BB′, tolyl), 7.79 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 8.7 (CH2), 9.1 (CH2), 12.4 (CH), 21.3 (2 × ArMe), 123.9 (CHR), 125.9, 126.3, 129.0, 130.4, 132.6, 141.3, 141.7, 142.3, 150.6 (CHS).
)-S-(2-Cyclopropylethenyl)-S-p-tolyl-N-p-tosylsulfimide..
δ
H(250 MHz; CDCl3) 0.58 (2H, m, CH2), 0.93 (2H, m, CH2), 1.53 (1H, m, CH), 2.35 (3H, s, Me tolyl), 2.38 (3H, s, Me tosyl), 6.01 (1H, dd, J = 10.8, 14.5 Hz, CHR), 6.02 (1H, d, J = 14.5 Hz, CHS), 7.16 (AA′ of AA′BB′, tolyl), 7.26 (AA′ of AA′BB′, tosyl), 7.49 (BB′ of AA′BB′, tolyl), 7.74 (BB′ of AA′BB′, tosyl); δC(62.9 MHz; CDCl3) 8.7 (2 × CH2), 14.5 (CH), 21.3 (2 × ArMe), 121.2 (CHR), 126.2, 126.5, 129.0, 130.4, 132.3, 141.3, 141.6, 142.7, 150.7 (CHS); m/z (FAB) 360 (100%, MH+), 190 (14), 123 (46).
(RS,SR)-S-[2-Phenyl-2-(phenylmethoxy)ethyl]-S-p-tolyl-N-p-tosylsulfimide 17a
A solution of vinyl sulfimide 16b (200 mg, 0.5 mmol) and sodium hydride (60%; hexane washed, 4 mg, 20 mol%) in benzyl alcohol (30 cm3) was stirred at room temperature for 3 days. Water (50 cm3) was added and then the aqueous solution was extracted with CH2Cl2 (3 × 20 cm3). The combined organic extracts were dried over magnesium sulfate and evaporated to dryness to leave a white residue (>95% de). Column chromatography on silica with 10% chloroform–ethyl acetate as eluant followed by recrystallisation from ether–petroleum ether yielded 17a as a white powder (165 mg, 64%), mp 146–148 °C; Rf 0.44 (10% chloroform–ethyl acetate) (Found: C, 68.83; H, 5.79; N, 2.77. C29H29NO3S2 requires C, 69.15; H, 5.80; N, 2.78%) (Found: 504.1670; MH+ requires 504.1669); νmax(Nujol)/cm−1 1283, 1140, 1116, 1090, 967, 725, 656; δH(400 MHz; CDCl3) 2.33 (3H, s, Me tolyl), 2.35 (3H, s, Me tosyl), 3.15 (dd, J = 10.7, 12.8 Hz, CHaS), 3.27 (1H, dd, J = 2.3, 12.8 Hz, CHbS), 4.05 (1H, AB, J = 10.5, CH2Ph), 4.20 (1H, AB, J = 10.5 Hz, CH2Ph), 4.84 (1H, dd, J = 2.3, 10.7 Hz, CHPh), 7.18 (2H, AA′ of AA′BB′, tolyl), 7.25 (2H, AA′ of AA′BB′, tosyl), 7.30 (10H, m, Ar), 7.60 (2H, BB′ of AA′BB′, tolyl), 7.81 (2H, BB′ of AA′BB′, tosyl); δC(100.6 MHz; CDCl3) 21.27 (Me tolyl), 21.34 (Me tosyl), 62.43 (CH2S), 70.99 (CH2Ph), 74.96 (CH), 125.89, 126.80, 126.52, 127.74, 127.81, 128.25, 128.74, 128.99, 129.13, 130.45, 132.10, 137.23, 137.98, 141.28, 141.50, 142.99; m/z (CI/NH3) 504 (35%, MH+).(RS,SR)-S-(2-Phenyl-2-ethoxyethyl)-S-p-tolyl-N-p-tosylsulfimide 17b
A solution of vinyl sulfimide 16b (200 mg, 0.5 mmol) and sodium hydride (60%; hexane washed, 4 mg, 20 mol%) in ethanol (40 cm3) was stirred at room temperature for 5 days. Water (40 cm3) was added and then the aqueous solution was extracted with CH2Cl2 (3 × 40 cm3). The combined organic extracts were dried over magnesium sulfate and evaporated to dryness to leave a white residue (80% de). Column chromatography on silica with ether–petroleum ether (8∶1) as eluant followed by recrystallisation from ether–petroleum ether (8∶1) yielded 17b as a white powder (165 mg, 73%), mp 127–129 °C; Rf 0.33 (ether–petroleum ether) (Found: C, 64.96; H, 6.04; N, 3.34. C24H27NO3S2 requires C, 65.28; H, 6.16; N, 3.17%); νmax(Nujol)/cm−1 1295, 1282, 1140, 1090, 969, 722, 656; δH(400 MHz; CDCl3) 1.07 (3H, dd, J = 7.0, 7.0 Hz), 2.33 (3H, s, Me tolyl), 2.35 (3H, s, Me tosyl), 2.96 (dq, J = 8.8, 7.0 Hz, CH2O), 3.05 (dd, J = 10.9, 12.9 Hz, CHaS), 3.19 (1H, dd, J = 2.3, 12.9 Hz, CHbS), 3.23 (1H, dq, J = 8.8, 7.0 Hz, CH2O), 4.60 (1H, dd, J = 2.3, 10.9 Hz, CHPh), 7.18 (2H, AA′ of AA′BB′, tolyl), 7.24 (2H, AA′ of AA′BB′, tosyl), 7.25 (5H, m, Ph), 7.60 (2H, BB′ of AA′BB′, tolyl), 7.82 (2H, BB′ of AA′BB′, tosyl); δC(100.6 MHz; CDCl3) 15.0 (Me), 21.2 (Me tolyl), 21.3 (Me tosyl), 62.3 (CH2S), 64.6 (CH2O), 74.6 (CH), 125.9, 126.3, 128.4, 128.7, 129.0, 130.4, 132.4, 138.5, 141.3, 141.4, 142.9; m/z (CI/NH3) 442 (12%, MH+).(RS,SR)-S-[2-(4-Methoxyphenyl)-2-ethoxyethyl]-S-p-tolyl-N-p-tosylsulfimide 17c
A solution of vinyl sulfimide 16a (50 mg, 0.12 mmol) and sodium hydride (60%; hexane washed, ≈1 mg) in ethanol (5 cm3) was stirred at room temperature for 5 days. Water (10 cm3) was added and then the aqueous solution was extracted with CH2Cl2 (3 × 10 cm3). The combined organic extracts were dried over magnesium sulfate and evaporated to dryness to leave a white residue (80% de). Column chromatography on silica with ethyl acetate–petroleum ether (2∶3) as eluant followed by recrystallisation from ethyl acetate–petroleum ether (2∶3) yielded a mixture of inseparable diastereoisomers 17c and 18c as a white gum (18 mg, 32%), Rf 0.33 (ethyl acetate–petroleum ether 2∶3); δH(for 17c only; 400 MHz; CDCl3) 1.05 (3H, dd, J = 7.0, 7.0 Hz), 2.33 (3H, s, Me tolyl), 2.35 (3H, s, Me tosyl), 2.92 (dq, J = 8.8, 7.0 Hz, CH2O), 3.04 (dd, J = 10.9, 12.8 Hz, CHaS), 3.17 (1H, dd, J = 1.8, 12.8 Hz, CHbS), 3.20 (1H, dq, J = 8.8, 7.0 Hz, CH2O), 3.76 (3H, s, MeO), 4.55 (1H, dd, J = 1.8, 10.9 Hz, CHAr), 6.83 (2H, AA′ of AA′BB′, MeOC6H4), 7.17 (2H, BB′ of AA′BB′, MeOC6H4), 7.18 (2H, AA′ of AA′BB′, tolyl), 7.25 (2H, AA′ of AA′BB′, tosyl), 7.60 (2H, BB′ of AA′BB′, tolyl), 7.81 (2H, BB′ of AA′BB′, tosyl).(RS,SR)-S-[2-Phenyl-2-(phenylmethylamino)ethyl]-S-p-tolyl-N-p-tosylsulfimide 17d
A solution of vinyl sulfimide 16b (20 mg, 0.05 mmol) and sodium hydride (60%; hexane washed, 0.5 mg, 20 mol%) in benzylamine (4 cm3) was stirred at room temperature for 24 h. Water (5 cm3) was added and then the aqueous solution was extracted with dichloromethane (3 × 4 cm3). The combined organic extracts were dried over magnesium sulfate and evaporated to dryness to leave a white residue (60% de). The major isomer was isolated by column chromatography on silica with 10% chloroform–ethyl acetate as eluant followed by recrystallisation from ether–petroleum ether to yield 17d as a white powder (16.1 mg, 63%), mp 146–148 °C; Rf 0.66 (10% EtOAc–CHCl3) (Found: C, 69.27; H, 6.07; N, 5.50. C29H30N2O2S2 requires C, 69.29; H, 6.02; N, 5.57%); νmax(Nujol)/cm−1 3321, 1283, 1140, 1116, 1090, 813, 725; δH(400 MHz; CDCl3) 2.25 (3H, s, Me tolyl), 2.35 (3H, s, Me tosyl), 3.13 (1H, dd, J = 10.5, 12.9 Hz, CHaS), 3.35 (1H, dd, J = 2.2, 12.9 Hz, CHbS), 4.06 (1H, AB, J = 10.5 Hz, CH2Ph), 4.27 (1H, AB, J = 10.5 Hz, CH2Ph), 4.81 (1H, dd, J = 2.2, 10.5 Hz, CHPh), 7.10 (2H, AA′ of AA′BB′, tolyl), 7.24 (2H, AA′ of AA′BB′, tosyl), 7.30 (10H, m, Ph), 7.66 (2H, BB′ of AA′BB′, tolyl), 7.84 (2H, BB′ of AA′BB′, tosyl); δC(100.6 MHz; CDCl3) 21.2 (Me tolyl), 21.3 (Me tosyl), 62.5 (CH2S), 71.0 (CH2Ph), 74.96 (CH), 125.90, 126.34, 126.89, 127.58, 127.77, 128.26, 128.96, 129.10, 130.45, 132.2, 134.04, 138.0, 139.14, 139.94, 141.12, 141.92; m/z (CI/NH3) 503 (3%, MH+), 310 (42), 210 (53), 188 (100).(RS,SR)-S-[2-(4-Chlorophenyl-2-(phenylmethylamino)ethyl]-S-p-tolyl-N-p-tosylsulfimide 17e
A solution of vinyl sulfimide 16c (21.5 mg, 0.05 mmol) and sodium hydride (60%; hexane washed, 0.5 mg, 20 mol%) in benzylamine (4 cm3) was stirred at room temperature for 24 h. Water (5 cm3) was added and then the aqueous solution was extracted with dichloromethane (3 × 4 cm3). The combined organic extracts were dried over magnesium sulfate and evaporated to dryness to leave a yellow oil (60% de). The major isomer was isolated by column chromatography on silica with 10% chloroform–ethyl acetate as eluant followed by recrystallisation from ether–petroleum ether to yield 17e as a yellow solid (18.2 mg, 57%); Rf 0.64 (CHCl3–EtOAc 4∶1) (Found: C, 64.24; H, 5.27; N, 5.26. C29H29ClN2O2S2 requires C, 64.85; H, 5.44; N, 5.21%); νmax(Nujol)/cm−1 3321, 1281, 1142, 1090, 813, 755, 736; δH(400 MHz; CDCl3) 2.33 (3H, s, Me tolyl), 2.35 (3H, s, Me tosyl), 2.92 (1H, AB, CH2Ph), 3.04 (1H, dd, J = 10.9, 12.8 Hz, CHaS), 3.18 (1H, dd, J = 2.4, 12.8 Hz, CHbS), 3.35 (1H, AB, CH2Ph), 4.62 (1H, dd, J = 2.4, 10.9 Hz, CHAr), 6.83 (2H, AA′ of AA′BB, p-ClC6H4), 7.17 (2H, BB′ of AA′BB′, p-ClC6H4), 7.18 (2H, AA′ of AA′BB′, tolyl), 7.24 (2H, AA′ of AA′BB′, tosyl), 7.35 (5H, m, Ar), 7.60 (2H, BB′ of AA′BB′, tolyl), 7.81 (2H, BB′ of AA′BB′, tosyl); δC(100.6 MHz; CDCl3) 21.2 (Me tolyl), 21.3 (Me tosyl), 62.45 (CH2S), 71.09 (CH2Ph), 75.06 (CH), 125.98, 126.84, 127.00, 127.67, 127.97, 128.00, 128.47, 128.96, 129.19, 130.49, 132.42, 134.08, 138.0, 139.98, 141.22, 143.12; m/z (CI/NH3) 538 (5%, MH+).General procedure for the reaction of vinyl sulfimides 16 with isopropylamine
Vinyl sulfimide 16 was stirred with a small amount of NaH in isopropylamine (15 cm3) under nitrogen at room temperature. The reaction was monitored by TLC. On completion (about 6 h) the excess amine was removed in vacuo. The remaining solid was extracted into ether (30 cm3) and washed with water (2 × 20 cm3), dried using magnesium sulfate and concentrated in vacuo yielding a yellow oil. The crude product was purified by column chromatography on silica gel (chloroform–ethyl acetate 4∶1).General procedure for the reaction of vinyl sulfimides 16 with diethylamine
Vinyl sulfimide 16 was stirred with a small amount of NaH in diethylamine (5 cm3) under nitrogen at room temperature. The reaction was refluxed (3 days), then the excess amine was removed in vacuo and the resulting white solid extracted into ether (10 cm3) and washed with water (2 × 10 cm3) and dried using magnesium sulfate. The ether layer was concentrated in vacuo to yield a brown solid. The product was purified by column chromatography on silica using chloroform–ethyl acetate 4∶1.Reaction of vinyl sulfimide 16e with ethanol
The experimental conditions for the synthesis of 17b, but with isopropyl sulfimide 16e in place of 16b, were used. The crude 1H NMR spectrum of the product mixture showed, by comparison with the literature data,26 that the major product was N-(1,1-dimethylprop-2-enyl)toluene-p-sulfonamide 22.4-Methyl-1-[2-(phenylmethoxy)-2-phenylethylsulfanyl]benzene 23a
A solution of 17a (8 mg, 0.016 mmol) and 10% palladium on carbon (1 mg) in degassed EtOAc (2 cm3) was stirred at room temperature under a positive pressure of hydrogen gas for 5 days. Filtration through Celite using EtOAc as eluant and concentration under reduced pressure afforded a yellow oil. Column chromatography on silica using 10% chloroform–ethyl acetate as eluant gave a pale yellow oil (3 mg, 56%); δH(400 MHz; CDCl3) 2.30 (3H, s, Me), 3.10 (1H, dd, J = 5.2, 13.4 Hz, CHaS), 3.35 (1H, dd, J = 8.0, 13.4 Hz, CHbS), 4.29 (1H, AB, J = 11.8 Hz, CH2Ph), 4.47 (1H, dd, J = 5.2, 8.0 Hz, CHPh), 4.48 (1H, AB, J = 11.8 Hz, CH2Ph), 7.04 (2H, AA′ of AA′BB′, tolyl), 7.21 (2H, BB′ of AA′BB′, tolyl), 7.30 (10H, m, Ar); δC(100.6 MHz; CDCl3) 20.95 (Me), 42.32 (CH2Ph), 70.75 (CH2S), 79.98 (CH), 126.82, 127.65, 127.83, 128.05, 128.21, 128.53, 129.60, 130.03, 132.74, 136.03, 137.96, 140.60; m/z (CI/NH3) 352 (100%, M + NH4+).4-Methyl-1-[2-(phenylmethylamino)-2-phenylethylsulfanyl]benzene 3d
A solution of 17d (8 mg, 0.016 mmol) and 10% palladium on carbon (1 mg) in degassed EtOAc (2 cm3) was stirred at room temperature under a positive pressure of hydrogen gas for 5 days. Filtration through Celite using EtOAc as eluant and concentration under reduced pressure afforded a yellow oil. Column chromatography on silica using 10% chloroform–ethyl acetate as eluant gave a pale yellow oil (3 mg, 56%); δH(400 MHz; CDCl3) 2.30 (3H, s, Me), 3.18 (1H, dd, J = 5.2, 13.4 Hz, CHaS), 3.42 (1H, dd, J = 8.0, 13.4 Hz, CHbS), 4.35 (1H, AB, J = 11.8 Hz, CH2Ph), 4.52 (1H, dd, J = 5.2, 8.0 Hz, CHPh), 4.68 (1H, AB, J = 11.8 Hz, CH2Ph), 7.04 (2H, AA′ of AA′BB′, tolyl), 7.21 (2H, BB′ of AA′BB′, tolyl), 7.30 (10H, m, Ar); δC(100.6 MHz; CDCl3) 21.21 (Me), 42.39 (CH2Ph), 70.80 (CH2S), 80.02 (CH), 126.89, 127.67, 127.93, 128.15, 128.26, 128.53, 129.66, 130.02, 132.76, 136.08, 17.99, 140.63; m/z (CI/NH3) 351 (100%, M + NH4+).Acknowledgements
We thank the EPSRC for studentships to C. P. R., S. M. R. and T. J. S. and the EPSRC Mass Spectrometry Service.References
- P. C. Taylor, Sulfur Rep., 1999, 21, 241 Search PubMed.
- For a review of particular relevance to this work see V. K. Aggarwal, A. Ali and M. P. Coogan, Tetrahedron, 1999, 55, 293 CrossRef CAS.
- A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS.
- S. G. Pyne, Sulfur Rep., 1999, 21, 281 Search PubMed.
- (a) H. Takada, Y. Nishibayashi, K. Ohe, S. Uemura, C. P. Baird, T. J. Sparey and P. C. Taylor, J. Org. Chem., 1997, 62, 6512 CrossRef CAS; (b) G. Celentano, S. Colonna, N. Gaggero and C. Richelmi, Chem. Commun., 1998, 701 RSC.
- C. P. Baird and P. C. Taylor, J. Chem. Soc., Chem. Commun., 1995, 893 RSC; C. P. Baird and P. C. Taylor, J. Chem. Soc., Perkin Trans. 1, 1998, 3399 RSC.
- (a) T. Yamamoto and M. Okawara, Chem. Lett., 1975, 581 CAS; (b) T. Yamamoto, M. Kakimoto and M. Okawara, Tetrahedron Lett., 1977, 1659 CrossRef CAS; (c) K. Ikeda, H. Satoh and T. Yamamoto, Org. Prep. Proced. Int., 1992, 24, 548 Search PubMed.
- A. J. Clark, S. Rooke, T. J. Sparey and P. C. Taylor, Tetrahedron Lett., 1996, 37, 909 CrossRef CAS.
- A. J. Clark , S. M. Rooke , T. J. Sparey and P. C. Taylor , unpublished results..
- J. L. G. Ruano, J. C. Carretero, M. C. Carreno, L. M. M. Cabrejas and A. Urbano, Pure. Appl. Chem., 1996, 68, 925 CAS; S. M. Proust and D. D. Ridley, Aust. J. Chem., 1984, 37, 1677 CAS.
- (a) C. Maignan and R. A. Raphael, Tetrahedron, 1983, 39, 3245 CrossRef CAS; (b) X. Ji, D. Vanderhelm, R. V. Williams and W. J. Ebey, Acta Crystallogr., Sect. B, 1989, 45, 93 CrossRef; (c) B. Ronan and H. B. Kagan, Tetrahedron: Asymmetry, 1991, 2, 75 CrossRef CAS.
- R. S. Glass, K. Reineke and M. Shanklin, J. Org. Chem., 1984, 49, 1527 CrossRef CAS.
- E. Bertotti, G. Luciani and F. Montanari, Gazz. Chim. Ital., 1959, 89, 1564 Search PubMed.
- O. De Lucchi and L. Pasquato, Tetrahedron, 1988, 44, 6755 CrossRef CAS.
- L. A. Paquette, R. E. Moerck, B. Harirchian and P. D. Magnus, J. Am. Chem. Soc., 1978, 100, 1597 CrossRef CAS.
- T. L. Gilchrist and C. J. Moody, Chem. Rev., 1977, 77, 409 CrossRef CAS.
- B. M. Trost and D. L. van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS.
- (a) T. Yamamoto, M. Kakimoto and M. Okawara, Bull. Chem. Soc. Jpn., 1979, 52, 841 CAS; (b) R. Annunziata, M. Cinquini, S. Colonna and F. Cozzi, J. Chem. Soc., Perkin Trans. 1, 1981, 3118 RSC.
- D. J. Cram, J. Day, D. R. Rayner, D. M. von Schrilz, D. J. Duchamp and D. C. Garwood, J. Am. Chem. Soc., 1970, 92, 7369 CrossRef CAS.
- D. Craig and N. J. Geach, Synlett, 1992, 299 CrossRef CAS.
- S. G. Pyne, R. Griffith and M. Edwards, Tetrahedron Lett., 1988, 29, 2089 CrossRef CAS.
- S. G. Pyne, P. Bloem, S. L. Chapman, C. E. Dixon and R. Griffith, J. Org. Chem., 1990, 55, 1086 CrossRef CAS; S. G. Pyne and B. Dikic, J. Org. Chem., 1990, 55, 1932 CrossRef CAS.
- (a) C. R. Johnson, K. Mori and A. Nakanishi, J. Org. Chem., 1979, 44, 2065 CrossRef CAS; (b) H. Takada, Y. Nishibayashi, K. Ohe and S. Uemura, Chem. Commun., 1998, 1557 RSC.
- K. A. Hellmund, S. G. Pyne, B. W. Skelton and A. H. White, Aust. J. Chem., 1992, 45, 1923 CAS see also A. D. Briggs, W. Clegg, M. R. J. Elsegood, C. S. Frampton and R. F. W. Jackson, Acta Crystallogr., Sect. C, 1998, 54, 1335 CrossRef.
- W. E. Truce and J. A. Simms, J. Am. Chem. Soc., 1956, 78, 2756 CrossRef CAS.
- R. G. Shea, J. N. Fitzner, J. E. Fankhauser, A. Spaltenstein, P. A. Carpino, R. M. Peevey, D. V. Pratt, B. J. Tenge and P. B. Hopkins, J. Org. Chem., 1986, 51, 5243 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |