Studies on pyrazines. Part 37.1 Synthesis of 6-propionylpteridine-2,4(1H,3H![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) )-dione and its 1- and/or 3-methyl derivatives from marine natural products
)-dione and its 1- and/or 3-methyl derivatives from marine natural products
Nobuhiro
Sato
* and
Shunsuke
Fukuya
Department of Chemistry, Yokohama City University, Yokohama, 236-0027, Japan
First published on 24th December 1999
Abstract
The synthesis of 1,3-dimethyl-6-propionylpteridine-2,4(1H,3H![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) )-dione is described, which is completed by the cross-coupling reaction of 6-bromolumazine with a 1-ethoxyprop-1-enyl tin compound in the presence of a palladium catalyst and copper iodide. Similarly, 1,3-demethylated, and 1- and 3-methyl derivatives are prepared from the corresponding bromolumazines which are obtained by cyclization of 6-bromo-3-acetylamino- or -3-methylamino-pyrazinecarbonitrile with methyl isocyanate or methyl chloroformate. In contrast, the synthesis of 6-propynyllumazine relies on the palladium-catalyzed cross-coupling reaction of 6-bromo-3-methylaminopyrazinecarbonitrile with propyne, yielding 6-acetonylpteridine on treatment with aqueous mercury(II) sulfate.
)-dione is described, which is completed by the cross-coupling reaction of 6-bromolumazine with a 1-ethoxyprop-1-enyl tin compound in the presence of a palladium catalyst and copper iodide. Similarly, 1,3-demethylated, and 1- and 3-methyl derivatives are prepared from the corresponding bromolumazines which are obtained by cyclization of 6-bromo-3-acetylamino- or -3-methylamino-pyrazinecarbonitrile with methyl isocyanate or methyl chloroformate. In contrast, the synthesis of 6-propynyllumazine relies on the palladium-catalyzed cross-coupling reaction of 6-bromo-3-methylaminopyrazinecarbonitrile with propyne, yielding 6-acetonylpteridine on treatment with aqueous mercury(II) sulfate.
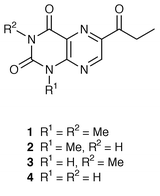
A series of lumazines having a propionyl, β-alkoxy- or β-hydroxy-propionyl group at C-6 were recently isolated from the metabolites of a swimming polychaete, Odontosyllis undecimdonta.2 This marine creature is commonly called fire worm since it luminesces during spawning at sunset in the middle of autumn at Toyama Bay, Japan. Because of the connection with the Odontosyllis bioluminescence or its biorhythm,3 these lumazine products are of considerable interest. Synthesis of 6-acyllumazines has been realized only by homolytic acylation of 1,3-dimethyllumazines,4 but we found some drawbacks in yield and regioselectivity of the radical-acylation products in the course of our existing work. A widely used procedure for synthesis of pteridines utilizes a manipulation of 3-aminopyrazinecarboxylic acid derivatives, and promises an unequivocal synthesis of the 6-substituted isomer.5 In this paper, we report on a program aimed at the synthesis of 6-propionylpteridine-2,4(1H,3H
)-diones 1–4, of which all except 2 are natural products, starting from 6-bromo-3-aminopyrazinecarbonitriles.
Most of the reported syntheses6,7 of pyrazinyl ketones involve treatment of a pyrazinecarboxylic ester or carbonitrile with an alkyllithium or Grignard reagent.6 An alternative method involves lithiation of pyrazines with an organolithium reagent followed by carbonylation, for example with N,N-dialkylcarboxamides.7 These routes, however, seem to be less suited for our synthesis because of the limited accessibility of the starting materials and the susceptibility of the pyrimidinedione ring to organometallic reagents, and so we needed some other synthetic strategy to meet our goal. The palladium-catalyzed cross-coupling reaction of halogenopyrazines with terminal acetylenes is an efficient method for synthesis of alkynylpyrazines,8–10 which are expected to be useful intermediates for the target acyllumazines since hydration of an acetylene is a known method for preparation of ketones.
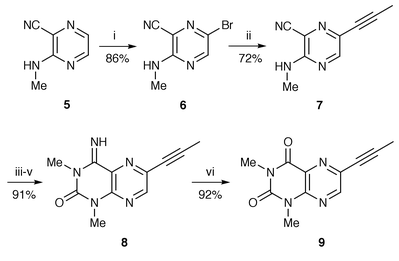
The key compound, 6-prop-1-ynyl-1,3-dimethylpteridine-2,4(1H,3H)-dione 9, was synthesized by a four-step sequence of reactions starting from 3-methylaminopyrazinecarbonitrile 511,12 (Scheme 1). Bromination of 5 was unexpectedly accomplished by the classical procedure, by treatment with bromine in aqueous acetic acid containing sodium acetate affording an 86% yield of bromopyrazine 6. Treatment of 5 with bromine in chloroform in the presence of pyridine or with N-bromosuccinimide (NBS) in aqueous dimethyl sulfoxide (DMSO), which greatly improved the bromination of aminopyrazines13 in comparison with the above traditional method, was ineffective in the present conversion to 6, and almost all of the aminopyrazine 5 was recovered. Cross-coupling of bromopyrazine 6 with propyne proceeded smoothly to form the alkynylpyrazine 7 in 72% yield; the success of this reaction was due to the use of bis(dibenzylideneacetone)palladium(0), Pd(dba)2, and particularly tri-o-tolylphosphine. Conversely, none of the desired product was obtained when tetrakis(triphenylphosphine)palladium(0)8,10 or dichlorobis(triphenylphosphine)palladium(II)9 was used as the catalyst.
 | ||
Scheme 1
Reagents and conditions: i, Br2, NaOAc, AcOH; ii, MeC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, Pd(dba)2, (o-tolyl)3P, CuI, Et3N, MeCN (reflux); iii, NaH, THF; iv, MeNCO; v, aq. HCl then NaHCO3; vi, aq. HCl, MeCN (reflux) then NaHCO3. CH, Pd(dba)2, (o-tolyl)3P, CuI, Et3N, MeCN (reflux); iii, NaH, THF; iv, MeNCO; v, aq. HCl then NaHCO3; vi, aq. HCl, MeCN (reflux) then NaHCO3.
| ||
It has been demonstrated that transformation of 3-methylaminopyrazinecarbonitrile 5 into pteridine derivatives is realized by treatment with sodium hydride in tetrahydrofuran (THF), followed by methyl isocyanate or methyl chloroformate.11 In our current synthesis, however, a reasonable improvement of the reaction conditions was required to optimize the cyclization of 6-alkynyl-3-methylaminopyrazinecarbonitrile 7. Thus, the previously published procedure using 0.3 equivalent of sodium hydride, and running for 19 h at room temperature led to the formation of 1,3-dimethyllumazine 8 in only 43% yield together with 10% recovery of the starting material. The best yield of 8 (91%) was obtained using 0.2 equivalent of sodium hydride and a reduction of the reaction time to 1 h. Hydrolysis of 8 was readily effected with refluxing 2 M hydrochloric acid for 5 h to provide pteridinedione 9 in 92% yield, whereas the reaction at room temperature11 induced no hydrolysis. 1-Methylpteridine-2,4-dione 11 was obtained by treatment of 7 with 2 equivalents of sodium hydride and methyl chloroformate followed by cyclization with basic hydrogen peroxide in 78% overall yield (Scheme 2). When 1 equivalent of sodium hydride was used, as in the literature procedure,11 the intermediate 11 was produced in 50% yield, with 34% recovery of starting material.
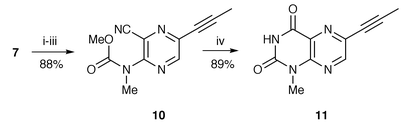 | ||
| Scheme 2 Reagents and conditions: i, NaH, THF; ii, ClCO2Me; iii, aq. HCl; iv, 30%H2O2, aq. NaOH, H2O, THF. | ||
The hydration of triple bonds is usually not regioselective.14 In this respect, the exclusive conversion of 2-alkynyl-3,5-diamino-6-chloropyrazines into the acylpyrazines, which was unexpectedly achieved by treatment with aqueous sodium sulfide and hydrochloric acid in methanol,15 was obviously relevant, and conceivably could be helpful in the final step of our synthetic approach. However, application of this method failed to synthesize the acyllumazine 1 and gave an almost quantitative recovery of the alkyne 9. In contrast, this substrate 9 was smoothly hydrated by mercury(II) sulfate in trifluoroacetic acid (TFA) containing a trace of water yielding the other possible isomer, acetonyllumazine 12, as the sole product (Scheme 3). Accordingly, we attempted to transfer the oxo group to the α position: i.e. conversion of 12 into 1. There are several procedures for such a transposition in the literature. We chose to use Corey’s methodology,16 consisting of C-nitrosation, reduction of the ketone, and finally dehydroxylation, with each step under mild reaction conditions. Surprisingly, reduction of the α-oximinoketone 13 with sodium borohydride led simultaneously to cleavage of the pyrimidinedione ring to yield pyrazinecarboxamide 15, and not the desired compound 14 (Scheme 3). The structure of 15 was indicated by the appearance of two N-methyl proton doublets at δ 2.81 and 2.96 in 1H NMR spectrum, and was clearly established by elementary analysis and the NMR spectra of the diacetyl derivative 16, formed only in the (Z) form.
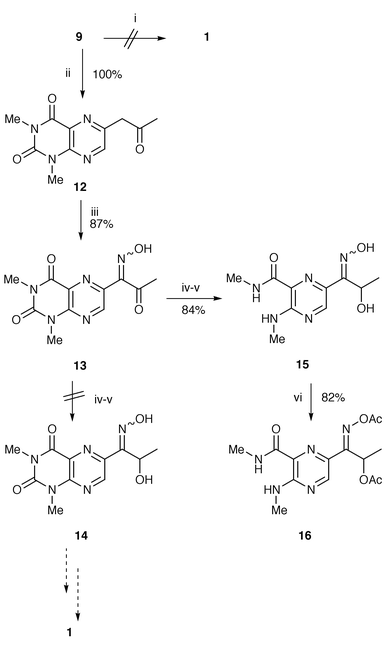 | ||
| Scheme 3 Reagents and conditions: i, Na2S, aq. HCl, MeOH (reflux); ii, HgSO4, H2O, TFA (reflux); iii, NaNO2, H2O, AcOH (0 °C); iv, NaBH4, aq. NaOH (rt); v, aq. HCl; vi, Ac2O, pyridine. | ||
Since the hydration of the ethynyl group reliably generates an acetyl derivative, utilization of acetyllumazine is still a possible protocol for synthesis of the propionyl compound 1. The trimethylsilylethynyllumazine 19, a precursor of acetyllumazine 20, was prepared as shown in Scheme 4, the sequence of reactions differing from those in the above synthesis of alkynyllumazine (Scheme 1), i.e. 6-bromolumazine 17 was initially constructed and subsequently cross-coupled with acetylene. The preparation of 17 and 18 from 6 was successfully realized using sodium hydride in the same way as the formation of 9 and 11, respectively, when the bromo substituent on the lumazines survived exposure to even 2 equivalents of the hydride. This synthetic method for 17 is much more convenient compared to the earlier one starting from 1,3-dimethyllumazine,17 since the use of 85% hydrogen peroxide is avoidable. The cross-coupling reaction of 17 with trimethylsilylacetylene proceeded easily in the presence of dichlorobis(triphenylphosphine)palladium(II) and copper(I) iodide in triethylamine leading to the almost quantitative formation of alkyne 19. Treatment of 19 with the mercury(II) ions provided directly the acetyl compound 20 in excellent yield. However, all attempts to effect the conversion of the acetyl into the propionyl group by base-promoted alkylation with methyl iodide were frustrated probably because the lumazine ring was decomposed by the strong base used: e.g. sodium hydride, lithium diisopropylamide, lithium hexamethyldisilazide or butyllithium, inferred from recovery of a trace of the starting material. Similarly, methylation via the enamine proved ineffective, and the acetyl compound 20 was almost completely recovered without forming the intermediate.
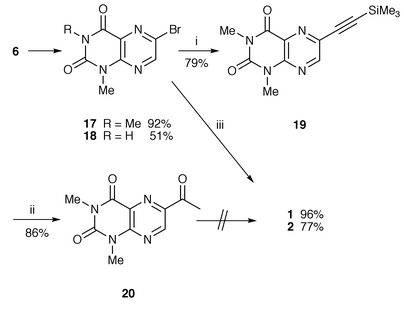 | ||
| Scheme 4
Reagents and conditions: i, Me3SiCCH, Pd(PPh3)2Cl2, CuI, Et3N (50–60 °C); ii, HgSO4, H2O, TFA; iii, 24, Pd(Ph3P)2Cl2, CuI, Et3N, MeCN (reflux).
| ||
A significant finding of the above unsuccessful project is the facile synthesis of 6-bromolumazine 17 and its high reactivity to palladium-catalyzed reaction so that we turned our attention to transition metal mediated acylation of bromolumazines to reach our goal. Since it is seemingly impossible to metallate the 6-bromolumazines due to the susceptibility of the lumazine ring to strong bases, an alternative method utilizing an organometallic acylation agent was explored. (1-Ethoxyvinyl)tributyltin was found to be effective as such a reagent, i.e. the organotin compound can couple with bromobenzenes in the presence of dichlorobis(triphenylphosphine)palladium(II) in toluene at 100 °C for 20 h yielding acetophenones.18 Under identical conditions, however, the acylation of 17 with (1-ethoxyprop-1-enyl)tributyltin 21, which was easily prepared from ethyl prop-1-enyl ether, did not go to completion, and a more difficult problem was the impossible isolation of ketone 1 from the unreacted starting substrate. Finally, when the reaction was carried out in acetonitrile containing 6 mol% of copper(I) iodide and triethylamine at reflux for 5 h, the cross-coupling was best effected affording a 96% yield of the desired product 1 (Scheme 4). Copper iodide stimulates the substitution reaction, because in the absence of the reagent the substrate was not completely consumed even after refluxing for 24 h. Acylation of bromolumazine 18 with organotin 21 similarly produced propionyllumazine 2 in 77% yield when 1 equivalent of copper iodide was employed.
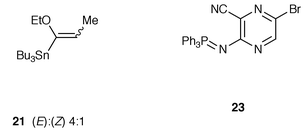
Our success in preparing 6-propionyllumazine 1 and 2 prompted us to examine the synthesis of the 1-demethylated derivatives 3 and 4. Compared to 3-methyllumazines 17 and 18, preparation of 6-bromolumazines 25 and 28 was much more troublesome, e.g. 3-amino-6-bromopyrazinecarbonitrile 22 failed to undergo cyclization with methyl isocyanate in the same way using sodium hydride or butyllithium as described above, and with recovery of 10–60% of the starting material. Instead a tandem aza-Wittig and cyclization methodology19 was attempted, but the iminophosphorane 23, whose synthesis was easily realized from 22 by a modified Kirsanov method using a triphenylphosphine–hexachloroethane–triethylamine system, resisted aza-Wittig reaction with methyl isocyanate and was completely recovered. However, the required bromolumazine 25 was successfully synthesized by the reaction sequence shown in Scheme 5. Acetylation of 22 in acetic anhydride containing 4-dimethylaminopyridine (DMAP) at >100 °C gave mainly the diacetylamino product. The desired monoacetylamino compound 24 was obtained in 40% yield under controlled condition using 1.3 equivalents of acetic anhydride in refluxing 1,2-dichloroethane, when 54% of the starting substrate was recovered. This acetyl compound 24 was treated with 0.6 equivalent of sodium hydride and then worked up in the same manner as in the synthesis of 3-methylated lumazine 17 to afford a 63% yield of lumazine 25. The amide 24 was also acylated with methyl chloroformate yielding imide 26, which was partially hydrolyzed during chromatographic work-up leading to carbamate 27 (Scheme 6). The mixture of 26 and 27 was treated with refluxing methanolic sodium methoxide to provide 4-methoxylumazine 29 in an overall yield of 86% from 24. Attempted cyclization of 26 or 27 with basic hydrogen peroxide gave a mixture of complex products and a trace of the slightly soluble bromolumazine 28, whose structure was inferred from its NMR spectrum. Acylation of bromo compounds 25 and 29 was accomplished by the same procedure for synthesis of propionyllumazines 1 and 2 to furnish the 1-demethylated products 3 and 4 in about 70% yield.
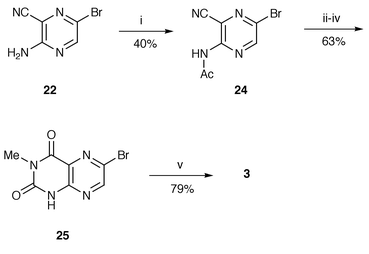 | ||
| Scheme 5 Reagents and conditions: i, Ac2O, ClCH2CH2Cl (reflux); ii, NaH, THF; iii, MeNCO; iv, aq. HCl (reflux); v, 24, Pd(Ph3P)2Cl2, CuI, Et3N, MeCN (reflux). | ||
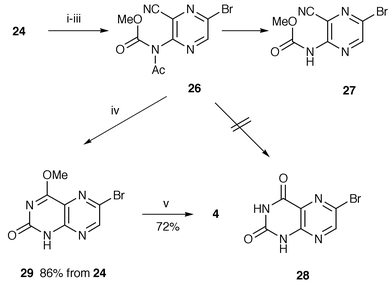 | ||
| Scheme 6 Reagents and conditions: i, NaH, THF; ii, ClCO2Me; iii, aq. HCl; iv, NaOMe, MeOH (reflux); v, 24, Pd(Ph3P)3Cl2, CuI, Et3N, MeCN (reflux). | ||
In conclusion, a convenient synthetic method for 6-bromolumazine was developed; the preparation of the 1-demethylated derivatives is noteworthy. These products could be versatile intermediates for other inaccessible lumazine compounds. As an example, the bromolumazines undergo palladium-catalyzed acylation with (1-ethoxyprop-1-enyl)tributyltin 21 affording propionyllumazines in good yields. We also found that copper iodide plays an important role in completing the cross-coupling reaction.
Experimental
General
Melting points were determined using a Büchi 535 or a Meltemp apparatus and are uncorrected. IR spectra were recorded on a Perkin Elmer Spectrum One. NMR spectra were obtained with a JEOL JNM EX270 (270 MHz 1H, 67.8 MHz 13C) and a Bruker Avance 400 (400 MHz 1H, 100.6 MHz 13C) instrument for solutions in CDCl3, unless otherwise noted, containing Me4Si as internal standard. J Values are given in Hz. Mass spectra were recorded on a JEOL JMS-AM II at 70 eV. Column chromatography was performed using Silica Gel 60. Drying refers to drying over MgSO4. Evaporation refers to evaporation under reduced pressure.3-N-Methylamino-6-bromopyrazinecarbonitrile 6
Cyanopyrazine 5 (1.070 g, 8.0 mmol) was added to a solution of NaOAc (1.969 g, 24 mmol) in AcOH (25 ml) containing water (1.3 ml), and to the mixture was added a solution of Br2 (1.9 g, 12 mmol) in AcOH (4 ml) over 10 min. The resulting mixture was stirred at rt for 4 h and then poured into ice–water. The aqueous solution was extracted with CHCl3 (4 × 50 ml), and the combined extracts were washed with 10% aqueous NaOH solution and then water. After drying, the solution was evaporated, and the residue was sublimed at 80 °C (2.5 mmHg) providing bromopyrazine 6 (1.472 g, 86%) as pale yellow needles, mp 140–141 °C (hexane–EtOAc, 6∶1) (Found: C, 33.8; H, 2.5; N, 26.3. C6H5N4Br requires C, 33.8; H, 2.4; N, 26.3%); νmax (KBr)/cm−1 3473, 3358 (NH), 2227 (CN), 1586, 1506, 1403, 1217 and 1149; δH 3.07 (3H, d, J 5.0, NCH3), 5.39 (1H, br s, NH) and 8.32 (1H, s, ArH); δC 28.3 (NCH3), 112.8 (Ar C), 114.3 (CN), 124.4 (Ar C), 149.1 (Ar CH) and 155.3 (Ar C).
3-N-Methylamino-6-(prop-1-ynyl)pyrazinecarbonitrile 7
A mixture of 6 (2.130 g, 10.0 mmol), Pd(dba)2 (0.290 g, 0.50 mmol), tri-o-tolylphosphine (0.304 g, 1.0 mmol) and CuI (95%, 0.095 g, 0.47 mmol) was purged by passage of argon after evacuation of air, and then dry MeCN (50 ml) and triethylamine (7.0 ml, 50 mmol) were added via a syringe. The mixture was refluxed with stirring and propyne (3.2 g, 80 mmol) was bubbled into it for 2 h. After being cooled, the mixture was filtered through Celite and evaporated. The residue was twice subjected to chromatography on silica (40 g × 2, hexane–EtOAc, 5∶1) affording the acetylene 7 (1.242 g, 72%) as pale yellow needles, mp 177–178 °C (hexane–EtOAc, 5∶1) (Found: C, 62.4; H, 4.55; N, 32.7. C9H8N4 requires C, 62.8; H, 4.7; N, 32.5%); νmax (KBr)/cm−1 3363 (NH), 2228 (CC and CN), 1594, 1517, 1336 and 1203; δH 2.08 (3H, s, CH3), 3.08 (3H, d, J 5.0, NCH3), 5.45 (1H, br s, NH) and 8.28 (1H, s, ArH); δC 4.4 (CH3), 28.1 (NCH3), 75.7 (CC), 88.4 (CC), 113.4 (Ar C), 114.7 (CN), 128.6 (Ar C), 149.1 (Ar CH) and 154.3 (Ar C).
1,3-Dimethyl-6-(prop-1-ynyl)pteridine-2,4(1H,3H)-dione 9
A mixture of NaH (60% dispersion in mineral oil, 16 mg, 0.4 mmol) and aminocyanopyrazine 7 (0.344 g, 2.0 mmol) was placed under argon, and dry THF (10 ml) was added via a syringe. The resulting wine-red mixture was stirred for 20 min at rt and then methyl isocyanate (0.4 ml, 6.8 mmol) was added dropwise. After stirring at rt for 1 h, the mixture was diluted with 2 M hydrochloric acid (20 ml), filtered, neutralized with NaHCO3 and extracted with CHCl3 (3 × 20 ml). After washing with water and drying, the solution was concentrated to dryness under reduced pressure. The residue was purified by chromatography on silica (40 g, hexane–EtOAc, 1∶1) to give 4-iminopteridinone 8 (0.419 g, 91%) as a pale yellow solid, δH 2.17 (3H, s, CH3), 3.58 (s, 3H, NCH3), 3.61 (3H, s, NCH3), 8.50 (1H, s, ArH) and 9.28 (1H, br s, NH). This product was partially hydrolyzed with time at rt or recrystallization work-up leading to lumazine 9.A mixture of the imine 8 (0.917 g, 4.0 mmol) in 2 M hydrochloric acid (13 ml) and MeCN (8 ml) was refluxed with stirring for 5 h. After being cooled, the precipitate was collected by filtration to give lumazine 9 (0.787 g, 85%) as pale yellow needles. The filtrate was neutralized with NaHCO3 and extracted with CHCl3 (3 × 20 ml). Drying the extract and removal of the solvent gave a second crop (0.065 g, 92% total yield), mp 258–259 °C (EtOH) (Found: C, 57.6; H, 3.95; N, 24.8. C11H10N4 requires C, 57.4; H, 4.4; N, 24.3%); νmax (KBr)/cm−1 2229 (CC), 1716, 1672 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O), 1538, 1496, 1450, 1327, 1206 and 750; δH 2.13 (3H, s, CH3), 3.54 (3H, s, NCH3), 3.71 (3H, s, NCH3) and 8.61 (1H, s, ArH); δC 4.6 (CH3), 29.1 (NCH3), 29.5 (NCH3), 76.2 (CC), 91.8 (CC), 127.0 (Ar C), 135.9 (Ar C), 146.1 (Ar C), 150.1 (Ar CH), 150.4 (2-CO) and 159.3 (4-CO); m/z (EI) 230 (100%, M+), 144 (65) and 118 (46).
O), 1538, 1496, 1450, 1327, 1206 and 750; δH 2.13 (3H, s, CH3), 3.54 (3H, s, NCH3), 3.71 (3H, s, NCH3) and 8.61 (1H, s, ArH); δC 4.6 (CH3), 29.1 (NCH3), 29.5 (NCH3), 76.2 (CC), 91.8 (CC), 127.0 (Ar C), 135.9 (Ar C), 146.1 (Ar C), 150.1 (Ar CH), 150.4 (2-CO) and 159.3 (4-CO); m/z (EI) 230 (100%, M+), 144 (65) and 118 (46).
3-(N-Methoxycarbonyl-N-methylamino)-6-(prop-1-ynyl)pyrazinecarbonitrile 10
A mixture of NaH (60%, 0.120 g, 3.0 mmol) and aminocyanopyrazine 7 (0.172 g, 1.0 mmol) was placed under argon, and dry THF (5.0 ml) was added via a syringe. The resulting wine-red mixture was stirred for 30 min at rt and then methyl chloroformate (0.12 ml, 1.5 mmol) was added dropwise. After stirring at rt for 7 h, the mixture was diluted with 2 M hydrochloric acid (10 ml) and extracted with CHCl3 (3 × 10 ml). The extract was washed with aqueous NaHCO3 and then water, dried and evaporated. The residue was subjected to chromatography on silica (20 g; hexane–EtOAc, 3∶1) to give carbamate 10 (0.203 g, 88%) as tiny needles, mp 117.5–118 °C (MeOH) (Found: C, 57.1; H, 4.4; N, 24.4. C11H10N4O4 requires C, 57.4; H, 4.4; N, 24.3%); νmax (KBr)/cm−1 2240 (CC and CN), 1746 (CO), 1466, 1440, 1297, 938 and 755; δH 2.15 (3H, s, CH3), 3.43 (3H, s, NCH3), 3.86 (3H, s, OCH3) and 8.55 (1H, s, ArH); δC 4.6 (CH3), 35.3 (NCH3), 53.8 (OCH3), 75.4 (CC), 93.4 (CC), 114.0 (CN), 127.0 (Ar C), 137.1 (Ar C), 147.6 (Ar CH), 151.3 (Ar C) and 154.3 (CO). The starting material was recovered from further elution (11 mg, 6%).
1-Methyl-6-prop-1-ynylpteridine-2,4(1H,3H)-dione 11
The carbamate 10 (0.230 g, 1.0 mmol) in a solution of hydrogen peroxide (30%, 1.0 ml), 0.5 M aqueous NaOH (3.2 ml), THF (1.0 ml) and water (1.6 ml) was stirred at rt for 2 h, and then adjusted to pH 5 with concentrated sulfuric acid. The precipitated product was collected by filtration to provide 11 (0.169 g, 78%) as pale yellow needles. The filtrate was evaporated and the residue was extracted with CHCl3–MeOH to afford a second crop (0.023 g, 89% total yield), mp 287.5–288 °C (EtOH) (Found: C, 55.6; H, 3.5; N, 25.9. C10H8N4 requires C, 55.6; H, 3.7; N, 25.9%); νmax (KBr)/cm−1 2232 (CC), 1722, 1703 (CO), 1537, 1496 and 1299; δH [(CD3)2SO] 2.15 (3H, s, CH3), 3.44 (3H, s, NCH3), 8.77 (1H, s, ArH) and 11.98 (1H, br s, NH); δC [(CD3)2SO] 3.8 (CH3), 28.1 (NCH3), 76.4 (CC), 90.3 (CC), 128.4 (Ar C), 133.2 (Ar C), 147.6 (Ar C), 149.2 (Ar CH), 149.8 (2-CO) and 159.2 (4-CO); m/z (EI) 216 (100%, M+), 144 (46) and 118 (30).
1,3-Dimethyl-6-acetonylpteridine-2,4(1H,3H)-dione 12
To a slurry of HgSO4 (0.265 g, 0.9 mmol) in TFA (45 ml) containing water (4 ml) was added alkyne 9 (1.032 g, 4.48 mmol), and the mixture was stirred under reflux for 1.5 h. After evaporation, the residue was diluted with water and extracted with CHCl3 (3 × 20 ml). The combined extracts were washed with aqueous NaHCO3 and then brine, dried and evaporated. The residue was purified by chromatography on silica (25 g; hexane–EtOAc, 2∶1) to provide ketone 12 (1.110 g, 100%) as golden tiny needles, mp 197–197.5 °C (MeOH) (Found: C, 53.3; H, 4.6; N, 22.3. C11H12N4O3: C, 53.2; H, 4.9; N, 22.6%); νmax (KBr)/cm−1 1720 (CO), 1673 (CO), 1547, 1504, 1336 and 750; δH 2.34 (3H, s, CH3), 3.55 (3H, s, NCH3), 3.73 (3H, s, NCH3), 4.18 (2H, s, CH2) and 8.58 (1H, s, ArH); δC 29.1 (NCH3), 29.4 (NCH3), 30.3 (CH3), 49.0 (CH2), 126.6 (Ar C), 145.9 (Ar C), 147.0 (Ar C), 148.9 (Ar CH), 150.5 (2-CO), 160.2 (4-CO) and 203.7 (CO); m/z (EI) 248 (15%, M+), 233 (6), 206 (100) and 120 (15).
1,3-Dimethyl-6-(1-hydroxyimino-2-oxopropyl)pteridine-2,4(1H,3H![[hair space]](https://www.rsc.org/images/entities/h3_char_200a.gif) )-dione 13
)-dione 13
A solution of NaNO2 (0.272 g, 4.0 mmol) in water (2.7 ml) was added dropwise to a stirred solution of acetonyllumazine 12 (0.495 g, 2.0 mmol) in AcOH (10 ml) at <5 °C, and the resulting mixture was stirred for 30 min and slowly warmed to rt over 1 h. After adding water, the solution was extracted with CHCl3 (4 × 30 ml) and the combined extracts were washed with aqueous NaHCO3, dried and evaporated. The residue was chromatographed on silica (25 g; hexane–EtOAc, 1∶1) affording oxime 13 (0.482 g, 87%) as light tan tiny needles, mp 188–193 °C (EtOH) (Found: C, 47.7; H, 4.1; N, 25.0. C11H11N5O4 requires C, 47.7; H, 4.0; N, 25.3%); νmax (KBr)/cm−1 3443, 1718 (CO), 1668 (CO), 1546, 1505, 1297 and 749; δH 2.59 (0.8 × 3H, s, CH3), 2.63 (0.2 × 3H, s, CH3), 3.53 (0.2 × 3H, s, NCH3), 3.56 (0.8 × 3H, s, NCH3), 3.74 (0.2 × 3H, s, NCH3), 3.76 (0.8 × 3H, s, NCH3), 9.07 (0.2H, s, ArH) and 9.18 (0.8H, s, ArH).
N-Methyl-(3-N′-methylamino)-6-(1-acetoxyimino-2-acetoxypropyl)pyrazinecarboxamide 16
Sodium borohydride (34 mg, 0.9 mmol) was added in portions to a stirred solution of oxime 13 (0.253 g, 0.91 mmol) in 0.5 M NaOH solution (10 ml) at ambient temperature, and the mixture was stirred for 1 h. After addition of 1 M hydrochloric acid (12 ml), the solution was extracted with EtOAc (3 × 15 ml), and the combined extracts were washed with brine, dried and evaporated to give pyrazinecarboxamide 15 (0.193 g, 84%). The crude product was dissolved in Ac2O (15 ml) containing pyridine (1.5 ml), and the mixture was stirred at rt for 3 h and then evaporated. The residual oil was diluted with EtOAc (20 ml) and the organic phase was washed with aqueous CuSO4 and brine, dried and evaporated. The residue was purified by chromatography on silica (7 g; hexane–EtOAc, 2∶1) to afford an oil, which was crystallized by ultrasonic work-up in hexane followed by evaporation to provide acetate 16 (0.210 g, 82%) as pale yellow tiny needles, mp 121–122 °C (hexane) (Found: C, 50.1, H, 5.7; N, 20.8. C14H19N5O5 requires C, 49.9; H, 5.7; N, 20.8%); νmax (KBr)/cm−1 3396 (NH), 1798, 1732 (CO), 1664 (CO), 1588, 1244, 1181 and 939; δH 1.59 (3H, d, J 6.6, CH3CH), 1.98 (3H, s, CH3CO), 2.27 (3H, s, CH3CO), 2.98 (3H, d, J 5.0, NCH3), 3.10 (3H, d, J 5.0, NCH3), 6.58 (1H, q, J 6.6, CH), 7.90 (1H, br s, NH), 9.03 (1H, s, ArH) and 9.13 (1H, br s, NH); δC 17.1 (CH3), 19.8 (CH3), 21.1 (CH3), 25.9 (NCH3), 27.5 (NCH3), 68.3 (CH), 125.6 (Ar C), 128.0 (Ar C), 150.5 (Ar CH), 154.3 (Ar C), 157.3 NCO), 166.2 (CN), 168.0 (CH3CO) and 170.9 (CH3CO).
1,3-Dimethyl-6-bromopteridine-2,4(1H,3H)-dione 17
This compound was prepared by the above procedure for the synthesis of alkynyllumazine 9. When bromopyrazine 6 (2.130 g, 10.0 mmol) was treated with NaH (60%, 0.080 g, 2.0 mmol) and methyl isocyanate (1.78 ml, 30 mmol) in THF (50 ml), the crude 4-iminolumazine (2.790 g) was obtained. Acidic hydrolysis gave the title compound 17 (2.506 g, 92%) as needles, mp 198–199 °C (EtOH) (lit.,17 mp 107–108 °C) (Found: C, 35.5; H, 2.4; N, 20.8. C8H7N4O2Br requires C, 35.45; H, 2.6; N, 20.7%); νmax (KBr)/cm−1 1719, 1667 (CO), 1537, 1479, 1167 and 751; δH 3.53 (3H, s, NCH3), 3.70 (3H, s, NCH3) and 8.71 (1H, s, ArH); δC 29.2 (NCH3), 29.7 (NCH3), 127.6 (Ar C), 134.1 (Ar C), 147.2 (Ar C), 150.2 (2-CO), 150.5 (Ar CH) and 158.7 (4-CO); m/z (EI) 272 (100%), 269 (100%, M+), 243 (23), 241 (22), 215 (30), 213 (30), 187 (77), 185 (75), 160 (56), 158 (57), 106 (39) and 79 (61).
1-Methyl-6-bromopteridine-2,4(1H,3H)-dione 18
This compound was prepared by the above procedure for the synthesis of 1-methyllumazine 11. Treatment of bromopyrazine 6 (0.320 g, 1.5 mmol) with NaH (60%, 0.115 g, 2.9 mmol) and methyl chloroformate (0.2 ml, 2.6 mmol) in THF (6 ml), and then flash chromatography (38 g; hexane–EtOAc 4∶1) afforded the carbamate (0.339 g, 83%). A mixture of the carbamate (0.701 g, 2.6 mmol) in 0.5 M NaOH (8.5 ml), THF (5 ml) and water (4.3 ml) was treated with hydrogen peroxide (30%, 2.4 ml) and worked up as described in the synthesis of 11 to yield the title compound 18 (0.341 g, 51%) as light tan tiny needles, mp 250–251.5 °C (MeOH) (Found: C, 32.9; H, 1.8; N, 21.45. C7H5N4O2Br requires C, 32.7; H, 2.0; N, 21.8%); νmax (KBr)/cm−1 3195, 3076 (NH), 1696 (CO), 1567, 1539, 1482, 1307, 1284 and 1163; δH 3.67 (3H, s, NCH3), 8.74 (1H, s, ArH) and 8.95 (1H, br s, NH); δC 29.2 (CH3), 128.4 (Ar C), 134.4 (Ar C), 148.6 (Ar C), 149.2 (2-CO), 150.8 (Ar CH) and 158.2 (4-CO); m/z (EI) 258 (98%), 256 (100, M+), 215 (22), 213 (22), 187 (43), 185 (44), 160 (41), 158 (43), 106 (32) and 79 (46).
6-(Trimethylsilylethynyl)-1,3-dimethylpteridine-2,4(1H,3H)-dione 19
A mixture of 17 (1.084 g, 4.0 mmol), Pd(PPh3)2Cl2 (0.140 g, 0.20 mmol), and CuI (95%, 0.038 g, 0.20 mmol) was placed under argon, and then dry triethylamine (13 ml) and ethynyltrimethylsilane (0.85 ml, 6.0 mmol) were added via a syringe. The mixture was heated at 50–60 °C with stirring for 0.5 h. After being cooled with ice–water, the precipitate was collected by filtration and the filtrate was evaporated. The residue was purified by chromatography on silica (30 g; hexane–EtOAc 1∶1). Recrystallization of the combined products gave alkyne 19 (0.887 g, 79%) as pale yellow prisms, mp 131–131.5 °C (hexane–MeOH) (Found: C, 54.0; H, 5.65; N, 19.3. C13H16N4O2Si requires C, 54.1; H, 5.6; N, 19.4%); νmax (KBr)/cm−1 2961, 1727, 1684 (CO), 1490, 1204 and 847; δH 0.24 [9H, s, Si(CH3)3], 3.47 (3H, s, NCH3), 3.66 (3H, s, NCH3) and 8.63 (1H, s, ArH); δC
−0.5 (SiCH3), 29.1 (NCH3), 29.5 (NCH3), 99.7 (CC), 100.4 (CC), 127.1 (Ar C), 135.1 (Ar C), 146.3 (Ar C), 150.3 (2-CO), 150.5 (Ar CH) and 159.1 (4-CO).
6-Acetyl-1,3-dimethylpteridine-2,4(1H,3H)-dione 20
The ethynyllumazine 19 (0.200 g, 0.69 mmol) was added to a vigorously stirring solution of HgSO4 (0.124 g, 0.42 mmol) in TFA (21 ml) containing water (2.1 ml), and the mixture was refluxed for 1 h. After evaporation, to the residue was added CHCl3, and the organic phase was washed with saturated NaHCO3 solution and then brine. Drying and evaporation gave ketone 20 (0.140 g, 86%) as light tan tiny needles, mp 203–204 °C (MeOH) (Found: 51.3; H, 4.3; N, 23.8. C10H10N4O3 requires C, 51.3; H, 4.3; N, 23.9%); νmax (KBr)/cm−1 1723 (CO), 1679 (CO), 1545, 1502, 1279, 1121 and 749; δH 2.82 (3H, s, CH3), 3.57 (3H, s, NCH3), 3.77 (3H, s, NCH3) and 9.29 (1H, s, ArH); δC 25.7 (CH3), 29.1 (NCH3), 29.8 (NCH3), 125.7 (Ar C), 143.5 (Ar C), 147.0 (Ar CH), 149.7 (Ar C), 150.3 (2-CO), 159.2 (4-CO) and 197.7 (CO); m/z (EI) 234 (98%, M+), 219 (35), 206 (100), 191 (89), 165 (75), 134 (46) and 107 (100).
6-Bromo-3-(triphenylphosphoranylidene)aminopyrazinecarbonitrile 23
Triethylamine (0.6 ml, 4.3 mmol) was added dropwise under argon to a stirred mixture of aminopyrazine 2213 (0.352 g, 1.8 mmol), hexachloroethane (0.627 g, 2.7 mmol) and triphenylphosphine (0.696 g, 2.7 mmol) in dry benzene (20 ml), and the resulting mixture was refluxed for 5 h. After being cooled, undissolved material was filtered off and the filtrate was evaporated. The residue was purified by chromatography on silica (30 g; hexane–EtOAc 4∶1) to give iminophosphorane 23 (0.771 g, 95%) as yellow tiny needles, mp 214–215 °C (EtOH) (Found: 60.0; H, 3.4; N, 12.1. C23H16N4PBr requires C, 60.15; H, 3.5; N, 12.2%); νmax (KBr)/cm−1 1528, 1453, 1435, 1109, 992, 884, 721 and 691; δH 7.45–7.52 (6H, m, PhH), 7.55–7.59 (3H, m, PhH), 7.76–7.84 (6H, m, PhH) and 7.92 (1H, s, ArH).
3-Acetylamino-6-bromopyrazinecarbonitrile 24
Acetic anhydride (0.065 ml, 0.7 mmol, 1.3 equivalents) was added to a stirred solution of aminopyrazine 22 (0.100 g, 0.51 mmol) and DMAP (7 mg) in 1,2-dichloroethane (4 ml), and the mixture was refluxed for 22 h. After being cooled, MeOH was added and the mixture was evaporated. The residue was chromatographed on silica (14 g; hexane–EtOAc 5∶1) to give recovery of the starting material as the first fraction (0.054 g, 54%). The next fraction (hexane–EtOAc 1∶1) afforded acetylamino compound 24 (0.048 g, 40%) as tiny needles, mp 192–193 °C (benzene) (Found: C, 35.2; H, 1.9; N, 23.2. C7H5N4OBr requires C, 34.9; H, 2.1; N, 23.2%); νmax (KBr)/cm−1 3239 (NH), 2239 (CN), 1687 (CO), 1493, 1422, 1344, 1120 and 915; δH 2.39 (3H, s, CH3), 7.83 (1H, br s, NH) and 8.63 (1H, s, ArH); δC 24.3 (CH3), 113.1 (CN), 128.3 (Ar C), 134.2 (Ar C), 147.9 (Ar C), 148.7 (Ar CH) and 168.1 (CO).
6-Bromo-3-methylpteridine-2,4(1H,3H)-dione 25
This compound was prepared by the above procedure for synthesis of 9. Thus, a solution of acetylaminopyrazine 24 (0.405 g, 1.7 mmol) in dry THF (12 ml) was treated with NaH (60%, 0.041 g, 1.0 mmol) followed by methyl isocyanate (0.3 ml, 5.1 mmol), and the resulting mixture was worked up as described above to give bromolumazine 25 (0.225 g, 63%) after chromatography on silica (45 g; hexane–EtOAc 4∶1), as tiny needles, mp 249–250 °C (benzene) (Found: 33.1; H, 1.7; N, 21.3. C7H5N4O2Br requires C, 32.7; H, 2.0; N, 21.8); νmax (KBr)/cm−1 3475 (NH), 3043, 1722 (CO), 1632, 1541, 1477, 1437, 1316 and 1175; δH 3.67 (3H, s, NCH3), 8.74 (1H, s, ArH) and 9.03 (1H, br s, NH); δC 29.2 (NCH3), 128.4 (Ar C), 134.5 (Ar C), 148.6 (Ar C), 149.2 (2-CO), 150.9 (Ar CH) and 158.2 (4-CO); m/z (EI) 258 (100%), 256 (98, M+), 215 (26), 213 (27), 187 (50), 185 (50), 160 (48), 158 (47), 106 (33) and 79 (48).
6-Bromo-4-methoxypteridin-2(1H)-one 29
To a solution of acetylaminopyrazine 24 (0.603 g, 2.5 mmol) in dry THF (20 ml) was added in portions NaH (60%, 0.150 g, 3.8 mmol), and the mixture was stirred at rt for 0.5 h. Methyl chloroformate (0.35 ml, 4.5 mmol) was added via a syringe, and the mixture was stirred for 2.5 h. After acidification with 2 M hydrochloric acid, the mixture was extracted with EtOAc (3 × 20 ml), and the combined extracts were washed with aqueous NaHCO3, dried and evaporated. The residue was chromatographed on silica (35 g; hexane–EtOAc 4∶1) to give a mixture of 26 and its deacetylated product 27 (0.704 g). These crude compounds were dissolved in dry MeOH (40 ml), and sodium methoxide (0.180 g, 3.2 mmol) was added. The solution was refluxed with stirring for 12 h and then cooled. The precipitated product was collected by filtration, and the filtrate was evaporated and purified by chromatography on silica (42 g; hexane–EtOAc 1∶1) to give methoxylumazine 29 (combined yield: 0.555 g, 86% overall yield from 24) as tiny needles, mp 226–228 °C (EtOH) (Found: C, 32.75; H, 1.8; N, 21.5. C7H5N4O2Br requires C, 32.7; H, 2.0; N, 21.8%); νmax (KBr)/cm−1 1672 (CO), 1604, 1458, 1395 and 1128; δH [(CD3)2SO] 4.05 (3H, s, OCH3), 8.90 (1H, s, ArH) and 12.35 (1H, br s, NH); δC [(CD3)2SO] 55.1 (OCH3), 122.6 (Ar C), 131.8 (Ar C), 149.0 (Ar C), 151.6 (Ar CH), 154.3 (2-CO) and 166.3 (4-C); m/z (EI) 258 (100%), 256 (100, M+), 228 (80), 226 (77), 200 (66), 198 (62), 177 (19), 150 (21), 82 (32) and 70 (75).
Palladium-catalyzed acylation of bromolumazines with organotin 21
C), 1464, 1137 and 1094; δH 0.67–1.0 (18H, m, CH3 and CH2), 1.19–1.67 (15H, m, CH2), 3.62 (0.8 × 2H, q, J 6.8, OCH2), 3.68 (0.2 × 2H, q, J 6.8, OCH2), 4.68 (0.2H, q, J 6.8, CCH) and 5.27 (0.8H, q, J 7.0, CCH).
1,3-Dimethyl-6-propionylpteridine-2,4(1H,3H)-dione 1
A mixture of bromolumazine 17 (0.268 g, 1.0 mmol), Pd(PPh3)2Cl2 (36 mg, 5.1 mmol, 5 mol%) and CuI (95%, 12 mg, 6.0 mmol, 6 mol%) was placed under argon, and MeCN (5.0 ml), triethylamine (0.7 ml, 5.0 mmol) and organotin 21 (1.026 g, 2.7 mmol, 2.7 equivalents) were added via a syringe. The mixture was stirred under reflux for 5 h and then 1.5 M hydrochloric acid (30 ml) was added to quench the reaction. After refluxing for 1 h, the solution was neutralized with NaHCO3, and extracted with EtOAc (3 × 15 ml). The combined extracts were dried and evaporated. The residue was chromatographed on silica (22 g; hexane–EtOAc 19∶1) to remove the tin compounds. Further elution (hexane–EtOAc 4∶1) gave acyllumazaine 1 as needles (0.236 g, 96%), mp 148–149 °C (EtOH) (lit.,4 mp 141–142 °C) (Found: C, 53.2; H, 4.9; N, 22.3. C11H12N4O3 requires C, 53.2; H, 4.9; N, 22.6%); νmax (KBr)/cm−1 2989 (CH), 2897 (CH), 1671 (CO), 1538, 1502, 1107 and 751; δH 1.24 (3H, t, J 7.3, CH3), 3.33 (2H, q, J 7.3, CH2), 3.57 (3H, s, NCH3), 3.76 (3H, s, NCH3) and 9.28 (1H, s, ArH); δC 7.5 (CH3), 29.1 (NCH3), 29.7 (CH2), 31.2 (NCH3), 125.7 (Ar C), 143.3 (Ar C), 147.0 (Ar CH), 149.7 (Ar C), 150.4 (2-CO), 159.2 (4-CO) and 200.3 (CO); m/z (EI) 248 (79%, M+), 221 (88), 219 (90), 193 (81), 191 (92), 165 (39), 134 (47), 107 (100) and 79 (69).
The following compounds were prepared by the above procedure except that 1 equivalent of CuI and 7–9 mol% of the palladium catalyst were used.
![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) )-dione 2..
This compound was obtained from 18 in 77% yield as tiny needles, mp 229.5–230.5 °C (benzene) (Found: C, 51.7; H, 4.2: N, 23.6. C10H10N4O3 requires C, 51.3; H, 4.3; N, 23.9%); νmax (KBr)/cm−1 3194 (NH), 3071 (CH), 1733 (CO), 1688 (CO), 1542 and 1493; δH [(CD3)2SO] 1.12 (3H, t, J 7.3, CH3), 3.17 (2H, q, J 7.3, CH2), 3.50 (3H, s, NCH3), 9.17 (1H, s, ArH) and 12.14 (1H, br s, NH); δC [(CD3)2SO] 7.6 (CH3), 28.5 (NCH3), 30.5 (CH2), 127.5 (Ar C), 141.8 (Ar C), 145.8 (Ar CH), 150.0 (Ar C), 151.1 (2-CO), 159.4 (4-CO) and 199.7 (CO); m/z (EI) 206 (14%, M − CO), 177 (6), 107 (20) and 57 (19).
)-dione 3..
This compound was obtained from 25 in 79% yield as tiny needles, mp 229.5–230.5 °C (benzene) (Found: C, 51.3; H, 4.1; N, 23.7. C10H10N4O3 requires C, 51.3; H, 4.3; N, 23.9%); νmax (KBr)/cm−1 3234 (NH), 3106 (CH), 1733 (CO), 1688 (CO), 1542 and 1494; δH [(CD3)2SO] 1.13 (3H, t, J 7.3, CH3), 3.16 (2H, q, J 7.3, CH2), 3.50 (3H, s, NCH3), 9.16 (1H, s, ArH) and 12.09 (1H, br s, NH); δC [(CD3)2SO] 7.5 (CH3), 28.4 (NCH3), 30.4 (CH2), 127.4 (Ar C), 141.7 (Ar C), 145.7 (Ar CH), 149.9 (Ar C), 151.0 (2-CO), 159.3 (4-CO) and 199.6 (CO); m/z (EI) 220 (8%, MH − CH3), 192 (100), 163 (42), 93 (41) and 57 (87).
)-dione 4..
This compound was obtained from 29 in 72% yield as tiny needles, mp 282–284 °C (decomp.) (hexane–EtOH) (Found: C, 49.2; H, 3.9; N, 25.1. C9H8N4O3 requires C, 49.1; H, 3.7; N, 25.45%); νmax (KBr)/cm−1 3542 (NH), 3455 (NH), 3189, 1738 (CO), 1704 (CO), 1564, 1356 and 1249; δH [(CD3)2SO] 1.12 (3H, t, J 7.3, CH3), 3.15 (2H, q, J 7.3, CH2), 9.08 (1H, s, ArH), 11.84 (1H, br s, NH) and 12.31 (1H, br s, NH); δC [(CD3)2SO] 7.6 (CH3), 30.3 (CH2), 126.4 (Ar C), 142.5 (Ar C), 146.6 (Ar C), 149.8 (Ar CH), 151.3 (2-CO), 160.3 (4-CO) and 199.7 (CO); m/z (EI) 220 (10%, M+) 192 (100), 163 (35), 137 (11) and 120 (19).
)-dione 2..
This compound was obtained from 18 in 77% yield as tiny needles, mp 229.5–230.5 °C (benzene) (Found: C, 51.7; H, 4.2: N, 23.6. C10H10N4O3 requires C, 51.3; H, 4.3; N, 23.9%); νmax (KBr)/cm−1 3194 (NH), 3071 (CH), 1733 (CO), 1688 (CO), 1542 and 1493; δH [(CD3)2SO] 1.12 (3H, t, J 7.3, CH3), 3.17 (2H, q, J 7.3, CH2), 3.50 (3H, s, NCH3), 9.17 (1H, s, ArH) and 12.14 (1H, br s, NH); δC [(CD3)2SO] 7.6 (CH3), 28.5 (NCH3), 30.5 (CH2), 127.5 (Ar C), 141.8 (Ar C), 145.8 (Ar CH), 150.0 (Ar C), 151.1 (2-CO), 159.4 (4-CO) and 199.7 (CO); m/z (EI) 206 (14%, M − CO), 177 (6), 107 (20) and 57 (19).
)-dione 3..
This compound was obtained from 25 in 79% yield as tiny needles, mp 229.5–230.5 °C (benzene) (Found: C, 51.3; H, 4.1; N, 23.7. C10H10N4O3 requires C, 51.3; H, 4.3; N, 23.9%); νmax (KBr)/cm−1 3234 (NH), 3106 (CH), 1733 (CO), 1688 (CO), 1542 and 1494; δH [(CD3)2SO] 1.13 (3H, t, J 7.3, CH3), 3.16 (2H, q, J 7.3, CH2), 3.50 (3H, s, NCH3), 9.16 (1H, s, ArH) and 12.09 (1H, br s, NH); δC [(CD3)2SO] 7.5 (CH3), 28.4 (NCH3), 30.4 (CH2), 127.4 (Ar C), 141.7 (Ar C), 145.7 (Ar CH), 149.9 (Ar C), 151.0 (2-CO), 159.3 (4-CO) and 199.6 (CO); m/z (EI) 220 (8%, MH − CH3), 192 (100), 163 (42), 93 (41) and 57 (87).
)-dione 4..
This compound was obtained from 29 in 72% yield as tiny needles, mp 282–284 °C (decomp.) (hexane–EtOH) (Found: C, 49.2; H, 3.9; N, 25.1. C9H8N4O3 requires C, 49.1; H, 3.7; N, 25.45%); νmax (KBr)/cm−1 3542 (NH), 3455 (NH), 3189, 1738 (CO), 1704 (CO), 1564, 1356 and 1249; δH [(CD3)2SO] 1.12 (3H, t, J 7.3, CH3), 3.15 (2H, q, J 7.3, CH2), 9.08 (1H, s, ArH), 11.84 (1H, br s, NH) and 12.31 (1H, br s, NH); δC [(CD3)2SO] 7.6 (CH3), 30.3 (CH2), 126.4 (Ar C), 142.5 (Ar C), 146.6 (Ar C), 149.8 (Ar CH), 151.3 (2-CO), 160.3 (4-CO) and 199.7 (CO); m/z (EI) 220 (10%, M+) 192 (100), 163 (35), 137 (11) and 120 (19).
Acknowledgements
Thanks are due to Mr Yoshinori Itsuki and Miss Kaori Matsumoto for their participation in the initial stage of this work.References
- Part 36 N. Sato and M. Ono , J. Heterocycl. Chem., Search PubMedsubmitted..
- S. Inoue, K. Okada, H. Tanino, H. Kakoi and N. Horii, Chem. Lett., 1990, 367 CAS; S. Inoue, K. Okada, H. Tanino, H. Kakoi, Y. Ohnishi and N. Horii, Chem. Lett., 1991, 563 CAS; H. Kakoi, H. Tanino, K. Okada and S. Inoue, Heterocycles, 1995, 41, 789 Search PubMed.
- H. Tanino, H. Takakura, H. Kakoi, K. Okada and S. Inoue, Heterocycles, 1996, 42, 125 Search PubMed.
- R. Baur, E. Kleiner and W. Pfleiderer, Liebigs Ann. Chem., 1984, 1798 Search PubMed.
- E. C. Taylor, K. L. Perlman, I. P. Sword, M. Séquin-Frey and P. A. Jacobi, J. Am. Chem. Soc., 1973, 95, 6407 CrossRef CAS; E. C. Taylor, K. L. Perlman, Y.-H. Kim, I. P. Sword and P. A. Jacobi, J. Am. Chem. Soc., 1973, 95, 6413 CrossRef CAS.
- W. Schwaiger, J. M. Cornelissen and J. P. Ward, Food Chem., 1984, 13, 225 CrossRef CAS and references cited therein..
- A. Turck, D. Trohay, L. Mojovic, N. Plé and G. Quéguiner, J. Organomet. Chem., 1991, 412, 301 CrossRef CAS.
- Y. Akita and A. Ohta, Heterocycles, 1982, 19, 329 Search PubMed.
- T. Sakamoto, M. Shiraiwa, Y. Kondo and H. Yamanaka, Synthesis, 1983, 312 CrossRef CAS.
- Y. Akita, A. Inoue and A. Ohta, Chem. Pharm. Bull., 1986, 34, 1447 Search PubMed.
- K. Tsuzuki and M. Tada, J. Heterocycl. Chem., 1986, 23, 1299 Search PubMed.
- N. Sato and N. Matsui, J. Heterocycl. Chem., 1992, 29, 1689 Search PubMed.
- N. Sato and R. Takeuchi, Synthesis, 1990, 659 CrossRef CAS.
- For example: S. Uemura, H. Miyoshi, K. Sohma and M. Okano, J. Chem. Soc., Chem. Commun., 1975, 548 RSC.
- M. J. Chapdelaine, P. J. Warwick and A. Shaw, J. Org. Chem., 1989, 54, 1218 CrossRef CAS.
- E. J. Corey and J. E. Richman, J. Am. Chem. Soc., 1970, 92, 5276 CrossRef CAS.
- H. Steppan, J. Hammer, R. Baur, R. Gottlieb and W. Pfleiderer, Liebigs Ann. Chem., 1982, 2135 Search PubMed; W. Pfleiderer and W. Hutzenlaub, Chem. Ber., 1973, 106, 3149 Search PubMed.
- M. Kosugi, T. Sumiya, Y. Obara, M. Suzuki, H. Sano and T. Migita, Bull. Chem. Soc. Jpn., 1987, 60, 767 CAS.
- T. Okawa, S. Eguchi and A. Kakehi, J. Chem. Soc., Perkin Trans. 1, 1996, 247 RSC.
- J. A. Soderquist and G. J.-H. Hsu, Organometallics, 1982, 1, 830 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |