The control of molecular self-association in spin-coated films of substituted phthalocyanines†
Bashir M. Hassan, Hong Li and Neil B. McKeown*
Department of Chemistry, University of Manchester, Manchester, UK M13 9PL, neil.mckeown@man.ac.uk
First published on UnassignedUnassigned22nd December 1999
Abstract
Uniform thin films are needed for the exploration and exploitation of the fascinating optical and electronic properties of the phthalocyanine (Pc) macrocycle. Spin-coating has been demonstrated to be a simple, fast and reproducible method of fabricating uniform films. However, solubility in a volatile organic solvent is a prerequisite for this technique and previous studies have shown that groups placed on the Pc macrocycle can have a profound effect on the nanoscale structure of the resulting film. In this paper we attempt to establish some fundamental structure–property relationships which determine the molecular packing within such films. Previous studies are reviewed and the film forming properties of a number of Pc derivatives are examined. In particular, the formation of films in which the Pc cores are isolated from each other by the use of novel, sterically crowded hexadeca-substituted Pcs is examined.
Since their discovery over seventy years ago, phthalocyanines (Pcs) have become one of the most studied of all organic functional materials. In addition to their use as blue and green colorants, Pcs are of increasing interest for applications in non-linear optics (including optical limitation), xerography (as photoconductors), optical data storage (as the laser absorption layer within recordable compact discs), molecular electronics, photodynamic cancer therapy, solar energy conversion, catalysis and as the active component of gas sensors.1,2 In order to optimise their potential utility in electronic and optical devices, it is necessary to fabricate Pcs as uniform thin films in which the nanoscale architecture and ordering can be reproducibly controlled. In addition to vacuum sublimation1,3 and Langmuir–Blodgett (LB) film formation,1,4 both of which have been thoroughly studied, spin-coating technology is a useful method of preparing uniform Pc films.5 This simple technique involves the rapid drying of a drop of Pc solution as it rotates and spreads under centrifugal force on a substrate revolving at high speeds (2–4 × 103 rpm). The thickness of the resultant film is controlled by the concentration of the solution, the volatility of the solvent and by the rate of rotation of the substrate. Spin-coating has the great advantage of ease and speed of fabrication which makes it attractive for the industrial-scale manufacture of devices. This is reflected in the numerous patents that cite spin-coating as the chosen method for film fabrication.6 For many purposes, the desired film is a composite of an insoluble Pc pigment dispersed in a soluble polymer matrix.7 However, this paper will concentrate on the control of the structure within spin-coated films derived purely from a single Pc component. In order to induce the required solubility in a suitably volatile solvent (e.g. chloroform), substitution of the Pc macrocycle is necessary. Solubilising groups include alkyl (–CnH2n+1) or alkoxy (–OCnH2n+1) side-chains placed at either the peripheral (2,3,9,10,16,17,23,24) or non-peripheral (1,4,8,11,15,18,23,24) benzo positions. Other types of substituents which have been used to enhance solubility are poly(aryl ether) dendritic wedges (e.g. [G-2] and [G-3]), oligo(oxyethylene) or aryloxy groups. In addition, it is possible to place groups at the axial sites of ions (e.g. Si4+) held in the central cavity of the Pc macrocycle. It is apparent that the number, type and position of the groups have a profound influence, not only on the solubility of the derivative, but also on the molecular-scale architecture of the resulting film. In this paper we attempt to collate the relationship between the structure of a Pc and the nanoscale architecture of its spin-coated film in terms of the steric and electronic effects of the substituents. The local order obtained in previous studies of Pc spin-coated films is reviewed, and the film forming properties of several previously synthesised Pc derivatives are assessed together with those of some novel Pc derivatives, whose structures encourage the attached alkyl side-chains to lie out of the plane of the Pc core.
A literature survey of spin-coated films derived from Pcs
Generally, it has been noted that the spin-coating of soluble Pc derivatives produces uniform optically clear films which are either glassy (i.e. lacking long-range crystalline ordering) or crystalline but with domains of too small a size to be observed using a polarising microscope.5 Annealing a film at a temperature above the Pc's melting point often increases the crystal domain size but can destroy the uniformity of thickness due to ‘de-wetting' of the substrate (i.e. the formation of droplets). In most cases the molecular arrangement within the spin-coated film is not dependent on the conditions used for deposition (solvent, spin rate) or any post-deposition treatment. In addition, the local arrangement of Pc molecules within spin-coated films is usually similar to the bulk material or that within films deposited by other means (e.g. the LB technique). Many soluble Pcs also exhibit a columnar mesophase1,8 and heating the film above the crystal–mesophase transition can improve the long-range ordering of the film without destroying its homogeneity.9,10 The self-ordering of a Pc material in a mesophase is often intimately related to the arrangement within the spin-coated film and it is likely that mesogenic Pcs form a liquid crystal phase, at least transiently, as the solvent evaporates during deposition.Four general types of Pc arrangement have been observed in spin-coated films and these are cofacial (face-to-face), herringbone, edge-to-edge and isolated (Fig. 1a–d). The visible absorption spectrum of the deposited films, of direct interest for many optical applications,6,11 indicates which type of molecular packing is present. Each of the four basic types of arrangement displays a characteristic visible absorption spectrum determined by the nature and extent of the intermolecular exciton coupling between the aromatic cores of neighbouring Pc molecules.1,12 The major absorption in the visible region of Pc is the Q-band and exciton coupling can perturb both the position and appearance of this band. The cofacial arrangement of Pc molecules results in a blue-shifted Q-band; a herringbone arrangement leads to strong Davydov splitting giving bands at both higher and lower wavelengths relative to the solution phase Q-band; edge-to-edge interactions result in a red-shifted Q-band; and finally, films containing isolated cores will possess an unperturbed spectrum similar to that obtained from a dilute solution of the Pc (Table 1).
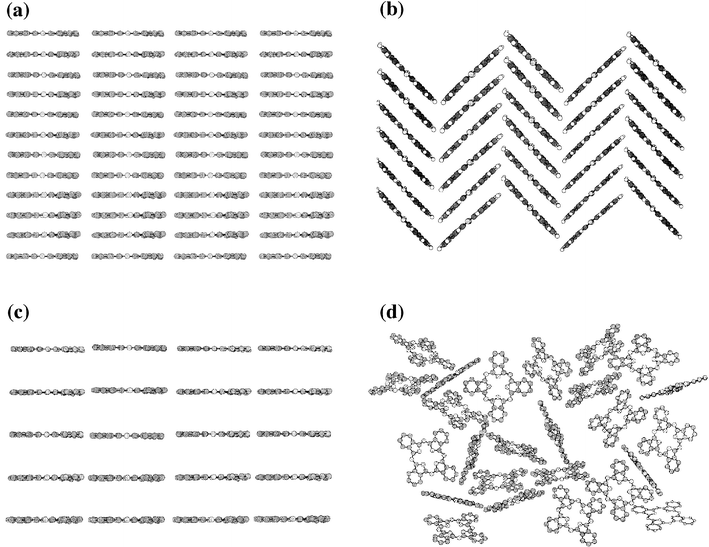 | ||
| Fig. 1 A diagrammatic representation of the four general types of Pc arrangement in spin-coated film: (a) cofacial or untilted columnar; (b) herringbone or tilted columnar; (b) edge-to-edge and (d) isolated. Note that only the Pc cores are shown and not the solubilising groups. In addition to the amorphous structure represented by (d), an ordered crystalline structure is also possible for the isolated arrangement in which the side-chains enforce isolation (e.g. Pc 18). | ||
| Pc | λmax (Qx, Qy) | (Qx + Qy)/2 | ΔQ | λmax (film) | Q-Band shift | Film type | Ref. |
|---|---|---|---|---|---|---|---|
| aMetal containing Pcs possess no Q-band split. | |||||||
| 1 | 700, 665 | 683 | 35 | 620 | −63 | C | 14 |
| 2a | 678 | — | — | 620 | −58 | C | 15 |
| 3 | 702, 664 | 683 | 38 | 622 | −61 | C | 18 |
| 4 | 715, 680 | 698 | 35 | 627 | −71 | C | 18 |
| 5 | 704, 668 | 686 | 36 | 625 | −61 | C | 10 |
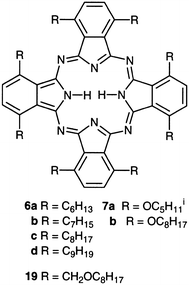
| 6a | 727, 695 | 711 | 32 | 738 | +27 | E | 19 |
| 6b–d | 727, 695 | 711 | 32 | 766, 634 | −77, +55 | H | 9 |
| 7a | 762, 740 | 751 | 22 | 777 | +26 | E (I) | 19 |
| 7b | 762, 740 | 751 | 22 | 855 | +104 | E | 19 |
| 8a | 680 | — | — | 692 | +12 | E | 22 |
| 9a | 680 | — | — | 688 | +8 | I (E) | 22 |
In order to interpret the structural information present in the visible absorption spectrum of a film derived from a substituted Pc, it is necessary to understand the effect of the solubilising groups on the position and appearance of the Q-band in the solution phase spectrum. The Q-band absorption has been assigned to a π–π* transition from the highest occupied molecular orbital (HOMO), of a1u symmetry, to the lowest unoccupied molecular orbital (LUMO) of eg symmetry. This results in a doubly degenerate first excited state of 1Eu symmetry. The split Q-band displayed by metal-free Pcs is due to its lower symmetry (D2h), as compared to that of metal-ion containing Pcs (D4h), and the consequent loss of degeneracy of the LUMO orbital to produce Qy and Qx states. Non-peripheral substitution, in particular, influences the position of the Q-band. For example, eight non-peripheral alkoxy moieties cause a 70 nm red-shift whereas alkyl side-chains in the same position result in a 30 nm red-shift relative to the Q-band of unsubstituted Pc (∼680 nm).13 In contrast, most peripheral substitution has only a modest effect on the Q-band position. Alkoxy chains in either peripheral or non-peripheral sites move the Soret absorption band from the UV region (∼350 nm) into the blue region (∼420 nm) resulting in the Pc possessing a green colour rather than the usual blue (cyan). An additional factor which affects the appearance of the Q-band of metal-free Pcs is the separation (ΔQ) of the Qx and Qy components which is steadily reduced as the Q-band is red-shifted. The reported Q-band parameters of the visible absorption spectrum of a number of Pcs both in solution and deposited as spin-coated film are given in Table 1.
In the crystalline phase Pc usually arranges itself in columnar stacks to maximise cofacial (i.e.
π–π) interactions.1 Additionally, this effect is observed in the strong tendency of Pcs to aggregate in concentrated solutions and accounts for the large number of Pc derivatives which form columnar liquid crystals often over a very wide temperature range.1,8 Tetra-substituted derivatives (e.g.1), tend to form spin-coated films in which the Pc molecules adopt an untilted cofacial arrangement (Fig. 1a) as shown by a broadened and blue-shifted Q-band.5,14 Unsurprisingly, spin-coated films of Pc derivatives which exhibit an untilted columnar mesophase at room temperature, such as those substituted with four or eight oligo(ethyleneoxy) substituents (e.g.2),15 or which form a solid anisotropic glass (e.g.3),16–18 in which the structure of the mesophase is frozen, also possess a cofacial arrangement. However, it is perhaps surprising that films composed of 4 also show this effect despite possessing four large dendritic substituents which were introduced with the aim of suppressing Pc self-association.17,18 In addition, the visible absorption spectra of spin-coated films prepared from amphiphilic Pc derivatives containing both alkyl and oligo(oxyethylene) side-chains (e.g.5) indicate a cofacial arrangement.10 Initially the films from 5 possess no long-range order, as indicated by X-ray diffraction, but self-organise into a lamellar structure (still with cofacial local ordering) when annealed at a temperature at which the Pc forms a columnar mesophase. This structure is analogous to the Y-type bilayer films obtained from the deposition of amphiphilic substances by the LB technique.

Highly uniform spin-coated films derived from most non-peripherally substituted octa-alkyl Pcs (6b–d) exhibit Davydov splitting of the Q-band in the visible spectrum.9 This effect originates from there being two translationally non-equivalent molecules in the crystallographic unit cell and suggests a herringbone arrangement (Fig. 1b) of the molecular stacks similar to that found in the β-crystal of unsubstituted metal-free Pc and many of its metal derivatives.1 Pc 6a exhibits anomalous behaviour as films derived from this compound exhibit a red-shifted Q-band indicating that edge-to-edge exciton interactions are the most significant.19 Unusually, 6a could be grown as crystals of sufficient size and quality for a single crystal X-ray diffraction analysis.20 This revealed an interesting configuration for the eight hexyl side-chains so that one is situated above the Pc ring, one below, and the remaining six are roughly in the same plane as the macrocycle resulting in a cofacial distance of over 8 Å (as compared to ∼3.5 Å in a cofacial stack) but with much closer edge-to-edge distances (Fig. 1c). A similar crystal structure is observed for 7a from which spin-coated films also display a red-shifted Q-band of similar appearance.19,21 Films derived from non-peripherally substituted unbranched octa-alkoxy Pcs (e.g.7b) demonstrate a highly red-shifted Q-band indicative of strong edge-to-edge interactions.19
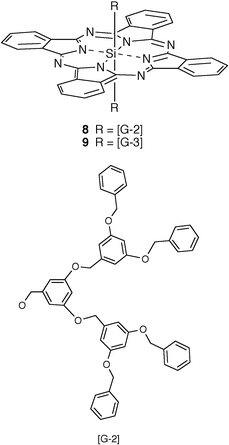
The axial substitution of Si4+Pc also encourages an edge-to-edge arrangement in the solid state. This is particularly well illustrated by 8 which has two second generation poly(aryl ether) dendritic groups as the axial substituents.18,22 This material is deposited as a glassy, optically clear film which displays a modest Q-band red-shift of 12 nm relative to the solution spectrum of 8. The red-shift is reduced to only 8 nm in spin-coated films of 9 which employs third generation dendritic wedges as axial substituents. The relatively weak exciton interactions within these films and their excellent clarity due to their non-scattering glassy structure (Fig. 1d) make them good approximations to ‘solid solutions' for various optical applications. A recent study has suggested another approach to the design of Pcs giving an isolated arrangement in spin-coated films due to severe steric crowding at the aromatic core. This involves placing a substituent at each of the 16 benzo subunits of the Pc (e.g.10).23

As part of an on-going synthetic programme at Manchester concerned with the construction of Pc-containing polymers, we have prepared a number of low molar mass Pc precursors, by-products and model compounds containing solubilising aryloxy (e.g.11) or alkyl side-chains (12–18). Therefore, it was of interest to determine the molecular arrangement within spin-coated films fabricated from these compounds and, specifically, whether an isolated arrangement of the Pc cores (Fig. 1d) is obtained from the novel derivatives 14–18. With the exception of 12 and 13, the solubilising groups are encouraged to lie out of the plane of the aromatic core due to either conformational effects (11), method of attachment to the Pc core (14 and 15) or severe steric crowding (16–18).

Experimental
Materials
The syntheses of Pcs 11,241225 and 1326 have been described previously. The full experimental details of the preparation of Pcs 14–18 will be given in subsequent publications.27 All Pcs gave satisfactory elemental analyses and spectroscopic data.
Film fabrication and characterisation
Spin-coated films of 11 and 14–18 were all prepared from chloroform solutions. Pcs 12 and 13 were only sparingly soluble in chloroform and films of these materials were deposited from warm solutions of toluene. The concentration for each solution was approximately 0.02 g ml−1. The substrate in each case was a clean, but otherwise untreated, glass microscope slide. Spin-coating was achieved, at 2000 rpm, using a Headway Research Inc. PM80 wafer spin cleaner. UV–visible spectra were recorded on a Shimadzu UV-260 spectrophotometer.Results and discussion
Pcs 11 and 14–18 gave spin-coated films of uniform appearance and excellent optical clarity. No birefringent crystalline domains were visible by polarising optical microscopy. In contrast, films derived from Pcs 12 and 13 appeared granular and uneven. Polarising microscopy confirmed the microcrystalline structure of these films. The poor film-forming properties of these two Pcs can be attributed to their comparatively low solubility which results in crystallisation as the solvent evaporates during deposition. The Q-band parameters of the visible absorption spectrum of Pcs 11–18 both in solution and as spin-coated film are given in Table 2. The colour of the film (green or blue) is determined by the position of the Soret band which is similar for solution and film spectra (Table 2).| Pc | λmax (Qx, Qy)a | Colour | (Qx + Qy)/2 | ΔQ | λmax (film) | Q-Band shift | Film type |
|---|---|---|---|---|---|---|---|
| aThe solution Soret adsorption band position is also given. The position of this band is similar in the film and determines its colour. bThe spin-coated films of 12 and 13 are of poor quality and their visible absorption spectra show broadening due to light scattering. | |||||||
| 11 | 704, 669, 343 | Blue | 687 | 35 | 625, | −59 | C |
| 12b | 704, 664, 425 | Green | 684 | 40 | 615, 760 | −69, 76 | H |
| 13b | 705, 665, 348 | Blue | 685 | 40 | 620, 740 | −65, 55 | H |
| 14 | 693, 653, 424 | Green | 673 | 40 | 699, 658 | +6 | I |
| 15 | 702, 666, 340 | Blue | 684 | 38 | 620 | −64 | C |
| 16 | 729, 696, 430 | Green | 713 | 33 | 736, 713 | +11 | I |
| 17 | 739, 711, 430 | Green | 725 | 28 | 744, 711 | +3 | I |
| 18 | 763, 735, 435 | Green | 749 | 28 | 767, 748 | +9 | I |
From the above survey it is reasonable to conclude that the degree of steric crowding at the Pc core, and hence the ability of the substituents to interfere with self-association, is the single most important factor to dictate the type of molecular packing arrangement adopted within a spin-coated film. Thus, for tetra-substituted Pcs even very large dendritic substituents (Pc 4) do not interfere with cofacial interactions. Compound 11, in which the aryloxy substituents should adopt a conformation to force the But moieties out of the plane of the Pc ring, is no exception and gives robust spin-coated films which display a cofacial arrangement (Fig. 1a; Table 2) despite showing no tendency to aggregate in solution.24
For spin-coated films of octa-substituted Pcs there is an interesting contest between π–π attractive interactions and steric effects which results in either a cofacial, herringbone, edge-to-edge or even an isolated arrangement depending on the position of the side-chains and their electronic influence on the Pc core. Thus, for 12 and 13, in which there is little steric obstruction to the side-chains resting in the same plane as the aromatic ring, a herringbone arrangement (Fig. 1b) is observed which is believed to maximise the attractive interactions between Pc cores (H-bonding and π–π interactions) as has been shown previously by molecular modelling.28 The exact crystalline structure of this tilted arrangement remains obscure due to the difficulty in obtaining crystals of sufficient size for a single crystal X-ray diffraction study, however, powder diffractions have been assigned to an orthorhombic structure for Pc 12.29
The electronic effect of the linking group between alkyl and macrocycle appears to be very influential in determining the local arrangement adopted by non-peripherally octa-substituted Pcs 6 and 7. For these compounds the primary steric interactions are those between alkyl groups situated on neighbouring benzo moieties which result in the displacement of some side-chains from the plane of the Pc ring as illustrated by the crystalline structures of 6a and 7a.20,21 Clearly this steric effect is very similar for both alkoxy and alkyl side-chains. Thus, the predominance of the herringbone packing displayed by members of the alkyl substituted series 6 (6a is an exception) appears related to stronger π–π interactions between the Pc cores than is the case with members of the series 7 which preferentially adopt an edge-to-edge arrangement. The effect of the linking group on the magnitude of π–π interactions can be rationalised by the theory of Hunter and Sanders.30 This theory suggests that the attractive interactions between the positively charged σ-bond skeleton and the negatively charged π-system are modulated by the repulsion between the similarly charged π-systems. Therefore, increasing the electron density of the aromatic system by the attachment of donating groups reduces the overall π–π interactions. The thermal behaviour of non-peripherally substituted Pcs appears to fit this model well as members of the series 19, containing electron-withdrawing alkoxymethyl (alkyl–O–CH2–) side-chains, display a columnar mesophase that is thermally stable up to the material decomposition temperature (>300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C).31 Members of series 6, with weakly electron-donating alkyl groups, display liquid crystallinity over a more modest thermal range32 and, remarkably, the series 7, with strongly electron-donating alkyloxy (alkyl–O–) linking groups, melt directly from the crystal to the isotropic liquid without the formation of any columnar mesophase and at moderate temperatures.19
°C).31 Members of series 6, with weakly electron-donating alkyl groups, display liquid crystallinity over a more modest thermal range32 and, remarkably, the series 7, with strongly electron-donating alkyloxy (alkyl–O–) linking groups, melt directly from the crystal to the isotropic liquid without the formation of any columnar mesophase and at moderate temperatures.19
The electronic effect of oxygen atoms attached to the Pc core may also explain the dramatic difference between the molecular arrangement observed in the spin-coated films of 14 (cofacial) and 15 (isolated). In these Pcs the alkyl groups are similarly attached via a five-membered ring which encourages them to lie out of the plane of the Pc core. Thus, the steric effect of the side-chains on core self-association is likely to be identical for both compounds. It appears that for 15 the moderating effect of the oxygen atoms on the π–π interactions, in combination with the side-chain configuration, is able to resist the tendency of the Pc cores to self-associate. The interesting thermal behaviour of these materials will be reported in a subsequent paper.27
The position and appearance of the Q-band absorption of films derived from 16–18 are very similar to those obtained from dilute solution (Table 2; Fig. 2) except for some broadening and a small red-shift (∼5–10 nm). Therefore, consistent with the previous study of compound 10,23 the severe steric crowding at the aromatic core of these hexadeca-substituted Pcs efficiently prohibits self-association of the aromatic cores in the solid phase. These three materials exhibit very different thermal behaviours: 16 is a low melting waxy solid, 17 is a liquid at room temperature and 18 is a highly crystalline material with a high melting point (202°C—compared with 68°C for Pc 7b). These properties result in films of 16 and 17 being relatively easily damaged but that of 18 is hard and robust. The structure within spin-coated films of 16 and 17 is likely to be well represented by the disordered arrangement depicted in Fig. 1d, however, that of 18 is expected to possess crystalline order, perhaps similar to that of 7a, but in which the 16 substituents prohibit significant cofacial and edge-to-edge interactions between neighbouring Pc cores. Further structural analysis of these materials is planned.
 | ||
| Fig. 2 The Q-band visible absorptions for (a) 16 as spin-coated film; (b) 16 in solution; (c) 17 as spin-coated film; (d) 17 in solution; (e) 18 as spin-coated film; (f) 18 in solution. | ||
To conclude, it is possible to prepare bespoke Pc-based spin-coated films with properties (colour, molecular association, optical absorption, mechanical resilience, etc.) tailored to the requirement of the intended application by using an appropriately substituted Pc derivative. Further modifications to the properties of the films could easily be made by placing a metal ion in the central cavity of the metal-free macrocycle. Importantly, it is possible to control the degree of molecular self-association. For a number of applications concerned with the electronic properties of the Pc macrocycle (e.g. use as an electronic sensor or photoconducting layer) a high level of molecular association is essential, whereas for many optical applications the ability to tune the position of the Q-band absorption and produce non-scattering films is paramount. It is hoped that the information presented in this paper will allow the appropriate choice of Pc derivative for use in a wide variety of film containing devices.
Acknowledgements
We thank the EPSRC (HL) and the Libyan Ministry for Higher Education (BMH) for financial support and Professor Mike Cook for providing data on Pcs 7a and 7b. Thanks are due to Saad Maksheed and, especially, the late Sarah Blake for help in acquiring visible absorption spectra.References
- N. B. McKeown, Phthalocyanine Materials: Synthesis, Structure and Function, Cambridge University Press, Cambridge, 1998 Search PubMed; N. B. McKeown, Chem. Ind., 1999, 92 Search PubMed.
- C. C. Leznoff and A. B. P. Lever, Phthalocyanines: Properties and Applications, Vol. 1–4, VCH, New York, 1989, 1993, 1993, 1996. Search PubMed.
- e.g. S. Dogo, J. P. Germain, C. Maleysson and A. Pauly, Thin Solid Films, 1992, 219, 244. Search PubMed.
- R. H. Tredgold, Order in Thin Organic Films, Cambridge University Press, Cambridge, 1994 Search PubMed; A. Ulman, Introduction to Ultrathin Organic Films, Academic Press, San Diego, 1991 Search PubMed; S. Baker, M. C. Petty, G. G. Roberts and M. V. Twigg, Thin Solid Films, 1983, 99, 53 Search PubMed; M. J. Cook, M. F. Daniel, K. J. Harrison, N. B. McKeown and A. J. Thompson, J. Chem. Soc., Chem. Commun., 1987, 1148 Search PubMed; J. D. Shutt, D. A. Batzel, R. V. Sudiwala, S. E. Rickert and M. E. Kenney, Langmuir, 1988, 4, 1240 CrossRef CAS; N. B. McKeown, M. J. Cook, A. J. Thomson, K. J. Harrison, M. F. Daniel, R. M. Richardson and S. J. Roser, Thin Solid Films, 1988, 159, 469 RSC; P. A. Albouy, J. Phys. Chem., 1994, 98, 8543 CrossRef CAS; M. Burghard, M. Schmelzer, S. Roth, P. Haisch and M. Hanack, Langmuir, 1994, 10, 4265 CrossRef CAS.
- M. Fujiki, H. Tabei and S. Imamura, Jpn. J. Appl. Phys., 1987, 26, 1224 CAS; S. M. Critchley, D. C. Davies, K. J. Markland and M. R. Willis, Synth. Met., 1991, 42, 1447 CrossRef CAS; S. M. Critchley and M. R. Willis, Int. J. Electron., 1994, 76, 809 CAS.
- M. Emmelius, G. Pawlowski and H. W. Vollman, Angew. Chem., Int. Ed. Engl., 1989, 28, 1445 CrossRef.
- e.g. N. Minami, K. Sasaki and K. Tsuda, J. Appl. Phys., 1983, 54, 6764. Search PubMed.
- J. Simon and C. Piechocki, J. Am. Chem. Soc., 1982, 104, 5245 CrossRef; S. Chandrasekhar, Liq. Cryst., 1993, 14, 3 CrossRef CAS; C. Destrade, P. Foucher, H. Gasparoux, N. H. Tinh, A. M. Levelut and J. Malthete, Mol. Cryst. Liq. Cryst., 1984, 106, 121 Search PubMed.
- S. M. Critchley, M. R. Willis, M. J. Cook, J. McMurdo and Y. Maruyama, J. Mater. Chem., 1992, 2, 157 RSC; S. M. Critchley, M. R. Willis, Y. Maruyama, S. Bandow, M. J. Cook and J. McMurdo, Mol. Cryst. Liq. Cryst., 1993, 230, 287 Search PubMed; M. J. Cook, D. A. Mayes and R. H. Poynter, J. Mater. Chem., 1995, 5, 2233 RSC.
- Z. Ali-Adib, G. J. Clarkson, N. B. McKeown, K. E. Treacher, A. S. Stennent and H. F. Gleeson, J. Mater. Chem., 1998, 8, 2371 RSC.
- G. de la Torre, P. Vázquez, F. Agulló-López and T. Torres, J. Mater. Chem., 1998, 8, 1671 RSC.
- E. S. Dodsworth, A. B. P. Lever, P. Seymour and C. C. Leznoff, J. Phys. Chem., 1985, 89, 5698 CrossRef CAS.
- M. J. Cook, A. J. Dunn, S. D. Howe, A. J. Thomson and K. J. Harrison, J. Chem. Soc., Perkin Trans. 1, 1988, 2453 RSC.
- S. Yanagi, T. Wada, J. Kumar, H. Sasabe and K. Sasaki, Mol. Cryst. Liq. Cryst., 1994, 255, 167 Search PubMed.
- N. B. McKeown and J. Painter, J. Mater. Chem., 1994, 4, 209 RSC; G. J. Clarkson, B. M. Hassan, D. R. Maloney and N. B. McKeown, Macromolecules, 1996, 29, 1854 CrossRef CAS; X. H. Wang, D. P. West, N. B. McKeown and T. A. King, J. Opt. Soc. Am. B, 1998, 15, 1895 Search PubMed.
- K. E. Treacher, G. J. Clarkson and N. B. McKeown, Liq. Cryst., 1995, 19, 887 CAS.
- M. Brewis, G. J. Clarkson, A. M. Holder and N. B. McKeown, Chem. Commun., 1998, 1979 RSC.
- N. B. McKeown, Adv. Mater., 1999, 11, 97 CrossRef CAS.
- M. J. Cook, J. Mater. Sci., Mater. Electron., 1994, 5, 117 CAS; M. J. Cook, J. Mater. Chem., 1996, 6, 677 RSC; M. J. Cook unpublished results. .
- I. Chambrier, M. J. Cook, M. Helliwell and A. K. Powell, J. Chem. Soc., Chem. Commun., 1992, 444 RSC.
- M. J. Cook, J. McMurdo and A. K. Powell, J. Chem. Soc., Chem. Commun., 1993, 903 RSC.
- M. Brewis, G. J. Clarkson, V. Goddard, M. Helliwell, A. M. Holder and N. B. McKeown, Angew. Chem., Int. Ed. Engl., 1998, 37, 1092 CrossRef CAS.
- M. Tian, T. Wada, H. Kimura-Suda and H. Sasabe, Mol. Cryst. Liq. Cryst., 1997, 294, 271 Search PubMed; M. Tian, T. Wada, H. Kimura-Suda and H. Sasabe, J. Mater. Chem., 1997, 7, 861 RSC; W. Eberhardt and M. Hanack, Synthesis, 1997, 95 CrossRef CAS.
- M. Brewis, G. J. Clarkson, P. Humberstone, S. Makhseed and N. B. McKeown, Chem. Eur. J., 1998, 4, 1633 CrossRef CAS.
- J. F. Van der Pol, E. Neeleman, J. W. Zwikker, R. J. M. Nolte, W. Drenth, J. Aerts, R. Visser and S. J. Picken, Liq. Cryst., 1989, 6, 577 CAS.
- G. J. Clarkson, N. B. McKeown and K. E. Treacher, J. Chem. Soc., Perkin Trans. 1, 1995, 1817 RSC.
- R. Black, B. Hassan, H. Li Makhseed, N. B. McKeown and N. Thompson, unpublished results. .
- P. Weber, D. Guillon and A. Skoulios, Liq. Cryst., 1991, 9, 369 CAS.
- K. Ohta, L. Jacquemin, C. Sirlin, L. Bosio and J. Simon, New J. Chem., 1988, 12, 751 Search PubMed.
- C. A. Hunter and J. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS.
- A. N. Cammidge, M. J. Cook, K. J. Harrison and N. B. McKeown, J. Chem. Soc., Perkin Trans. 1, 1991, 3053 RSC; A. N. Cammidge, M. J. Cook, S. D. Haslam, R. M. Richardson and K. J. Harrison, Liq. Cryst., 1993, 14, 1847 CAS.
- M. J. Cook, M. F. Daniel, K. J. Harrison, N. B. McKeown and A. J. Thomson, J. Chem. Soc., Chem. Commun., 1987, 1086 RSC; A. S. Cherodian, A. N. Davies, R. M. Richardson, M. J. Cook, N. B. McKeown, A. J. Thomson, J. Feinjoo, G. Ungar and K. J. Harrison, Mol. Cryst. Liq. Cryst., 1991, 196, 103 Search PubMed.
Footnote |
| † Basis of a presentation given at Materials Chemistry Discussion No. 2, 13–15 September 1999, University of Nottingham, UK. |
| This journal is © The Royal Society of Chemistry 2000 |