Structure and conductivity in LB films of low-dimensional polymer electrolytes†
Yungui Zhenga, Alain Gibaudb, Neil Cowlamc, Tim H. Richardsonc, G. Ungara and Peter V. Wright*a
aDept. of Engineering Materials, University of Sheffield, Mappin St., Sheffield, UK S1 3JD
bLaboratoire P.E.C. Rayons-X, Faculté des Science, Université du Maine, 72017, Le Mans Cedex, France
cDept. of Physics, University of Sheffield, Hounsfield Road, Sheffield, UK S3 7RH
First published on UnassignedUnassigned22nd December 1999
Abstract
LB films and bulk samples of complexes of (CF3SO2)2NLi and LiClO4 with polymers CH2OCH2- [(C6H3OR)CH2O(CH2OCH2)n−1] where R = –C16H33 or –C18H37 and n = 5 have been compared. Three-component systems incorporating equimolar proportions to the repeating unit of ‘expander' C16H33OH or C18H38 are also discussed. The laminar structures of the complexes have been modelled by molecular dynamics. The LB multilayer films (400–600 Å) have been characterised using surface pressure–area isotherms, low angle X-ray scattering and impedance spectroscopy. Conductivity measurements both in the plane of the films and normal to the plane are reported and compared with measurements on bulk (100 μm) films. Temperature-independent conductivity is observed in the ‘normal-to-plane' LB measurements. We propose that a morphological transformation occurs at the first heating during which the ion-mobile interlamellar planes become oriented normal to the electrodes.
Introduction
Conventional, solvent-free polymer electrolytes for transference of alkali metal ions (usually Li+) are generally based on amorphous polyethyleneoxy–salt ((EO)n–M+A−) mixtures but are widely assumed to have reached limits in conductivity (10−5–10−4 S cm−1 at 20–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C and ca. 10−3 S cm−1 at temperatures ≥ 80°C) which fall short of that required (ca. 10−3 S cm−1) for the successful operation of lithium batteries at temperatures below ca. 80°C. Suggestions for improvements in performance include the dissolution of low proportions of polymer in low glass-transition mixtures of lithium salts1 (‘polymer-in-salt' systems) or the incorporation of nanoparticulate fillers into the conventional systems.2
°C and ca. 10−3 S cm−1 at temperatures ≥ 80°C) which fall short of that required (ca. 10−3 S cm−1) for the successful operation of lithium batteries at temperatures below ca. 80°C. Suggestions for improvements in performance include the dissolution of low proportions of polymer in low glass-transition mixtures of lithium salts1 (‘polymer-in-salt' systems) or the incorporation of nanoparticulate fillers into the conventional systems.2We have proposed that low-dimensional systems may also offer a way forward by providing low impedance channels or planes for ion mobility and, if the ionic material is confined to monolayers, by inhibiting the formation of ion aggregates which suppress ion mobility in organic media of low dielectric constant. Other possible benefits relating to ion destabilisation are discussed below. However, like the ceramic fast-ion conductors which they emulate, low dimensional systems will require the conductive planes or channels to be favourably oriented approximately normal to the electrodes in order to optimise ion mobility between them. It is with this aspect of the structure and conductivity of some LB films of a low-dimensional polymer electrolyte system that this paper is principally concerned.
In recent years we have been studying organised, amphiphilic low-dimensional polymer electrolyte systems based upon polyethers of the general formula 1.3–13 When these molecules are fully organised the side groups R, which are generally long n-alkyl chains, condense together to create an ionophobic layer causing the polyether segments to generate a helical substructure. Alkali metal cations may be inserted into the helical tubes whilst the anions occupy the spaces between the helices. This is shown in the molecular dynamics model of Fig. 1 (which is a new representation for this paper). When n = 5 the repeating unit of 1 imitates the 18-bond helical repeat of the (EO)3–M+A− complexes14–18 (with a ‘dummy' site at the aromatic ring) and for R = −C16H33 the polymer is code-named ‘C16O5'. Although polymers with other ‘n' and side groups, R, have been prepared,19,20 C16O5 and its equimolar complexes with LiClO4, LiBF4, LiCF3SO3 and NaCF3SO3 have so far received most attention. These complexes are abbreviated, for example, C16O5∶LiClO4 (1∶1).
 | ||
| Fig. 1 Molecular dynamics model of C16O5∶LiClO4 (1∶1). One of 100 molecular dynamics ‘snapshots' at 20°C after ‘annealing' at 327°C. Red spheres: oxygens, pink spheres: lithium ions, large blue spheres: anions, small spheres are carbons and hydrogens. (Top) Side view of helices, (bottom) end view of helices. | ||
The polymers 1 are readily synthesised by standard procedures and molar masses of more than 105 have been obtained. Salt complexes may be readily prepared by co-dissolving the polymer and salt in acetone but following removal of the solvent a period of heat treatment at ca. 60°C is required to ensure that the salt is homogeneously distributed throughout the low-dimensional system.
The above two-component complexes have been developed further7,11,12 by incorporating alkane or substituted alkane molecules, RX, of appropriate length into the C16O5∶salt system. Such additives include the single stems C18H38 and C16H33OH or the double stem (C16H33O)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O which form equimolar three-component complexes with C16O5 and the lithium salt, denoted C16O5∶RX∶LiA (1∶1∶1). The RX component facilitates organisation of the amphiphilic system but also ‘expands' the internal ionophilic surfaces available for ion migration. The two- and three-component C16O5 systems incorporating LiClO4, LiBF4, LiCF3SO3 or NaCF3SO3 investigated so far are decidedly two-dimensional in both crystalline and liquid crystalline phases. Smectic A lc structures in bulk samples3–8 above the side chain melting temperatures (40–42°C) are readily identified by X-ray scattering and optical microscopy. Interestingly, it is the salt complexed with the polyether helical component which imparts the liquid crystalline character since the salt-free polymers become isotropic above side-chain melting.
O which form equimolar three-component complexes with C16O5 and the lithium salt, denoted C16O5∶RX∶LiA (1∶1∶1). The RX component facilitates organisation of the amphiphilic system but also ‘expands' the internal ionophilic surfaces available for ion migration. The two- and three-component C16O5 systems incorporating LiClO4, LiBF4, LiCF3SO3 or NaCF3SO3 investigated so far are decidedly two-dimensional in both crystalline and liquid crystalline phases. Smectic A lc structures in bulk samples3–8 above the side chain melting temperatures (40–42°C) are readily identified by X-ray scattering and optical microscopy. Interestingly, it is the salt complexed with the polyether helical component which imparts the liquid crystalline character since the salt-free polymers become isotropic above side-chain melting.
Assuming that ion mobility is enhanced by ion-destabilisation (consistent with the salt remaining in-phase) the ‘conventional' amorphous systems, although suppressing large-scale crystallinity, in general offer little constraint on the optimum stabilisation of cations by ‘wrap-around' segmental conformations. In ethoxy segments these generally involve trans (t)–gauche (g)–trans sequences in O–C–C–O bonds respectively. In such systems ion mobility is closely coupled to the matrix relaxation. Furthermore, amorphous systems should offer little impediment to the formation of ionic aggregates MA, M2A+, MA2−, M2A2,… in the low permittivity media by permitting their development and ion interchange with three-dimensional accessibility. In addition, the free volume required for the successful translation of an ion between coordinated sites contracts with reducing temperature bringing about an inevitable reduction in the conductivity.
Low-dimensional systems however, notwithstanding the inevitable constraint of ensuring that the planes or channels of optimum conductivity are favorably oriented between the electrodes, should by appropriate molecular design be capable of moderating the stabilities of ions in their interactions both with the polymer and with each other. With ions disposed at ‘internal surfaces' separated by ‘ionophobic barriers' three-dimensional nucleation of aggregates is precluded and the ions assume higher energies of complexed surfaces rather than those of ions within coulombic lattices. Furthermore, unimpeded or low-barrier pathways can in principle be constructed which may remain dilated on cooling for optimum low temperature conductivity.
These principles are in operation in the two-dimensional ceramic fast ion conductors such as sodium–β-alumina in which conductivities in excess of 10−2 S cm−1 at ambient temperatures may be achieved. The transposition of these principles to polymer electrolytes without loss of the inherent attractive features of the conventional systems is desirable. These include mechanical flexibility and compliance with electrode interfaces in electrochemical devices.
The schematic diagram of the two-dimensional systems under consideration shows ions complexed at internal surfaces defined by two ethoxy helices separated by ‘barriers' of alkyl material. As Fig. 1 shows, the stability of the cations is moderated by the ethoxy sequences along the helices which incorporate a dummy site at the phenyl ring which will have a lower interaction energy than the ether oxygens. The new representation of the lithium coordination (Fig. 1) is discussed below in more detail. The helix itself should provide a tube along which cations can drift with low hopping barriers between the adjacent oxygens whereas the spaces between the helices are essentially unimpeded channels along which anions may drift. The schematic (Fig. 2) suggests that ionic mobility should increase along the sequence a ≪ c < b. In the a direction the ionophobic barrier layers are encountered whereas b is the helical direction in which the cation can move smoothly between oxygen sites in cooperation with the anions. For mobility in the c direction the cations must hop relatively large distances (6–12 Å) between nearest helices and the anions must probably negotiate a zig-zag path between the layers.
 | ||
| Fig. 2 Schematic diagram of a three-component complex C16O5∶RX∶LiA (1∶1∶1). Both single- and double-stemmed n-alkyls are shown. • = functional group, X, A = anion. | ||
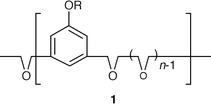
The bulk conductivities of the two and three-component systems which have been observed so far indicate that this overall strategy has substance notwithstanding the low proportion of ionophilic material. Fig. 3 shows conductivity data for two and three-component systems with LiClO4. Both types of system achieve ca. 10−2 S cm−1 at 100°C but the equimolar three-component systems such as C16O5∶C16H33OH∶LiClO4 (1∶1∶1) and C16O5∶C12H25OC6H5∶LiClO4 (1∶1∶1) have higher conductivities at temperatures closer to ambient. This may be attributed to the expansion of the internal surface area available to the ions by the RX component which both facilitates mobility and promotes molecular organisation. As we have shown,4–8 conductivity in these systems increases dramatically with organisation of the lamellar liquid-crystal structure as isolated pockets of ions are removed. The most highly organised bulk samples that we have so far been able to prepare have higher conductivities than amorphous PEO complexes at temperatures >ca. 50°C (above side chain melting), as Fig. 3 demonstrates. However, after side chain melting, a second transition is apparent in the heating cycles above ca. 60°C. This we tentatively attribute to disorganisation (expansion) of the interlamellar space occupied by the anions. The failure of lamellae to re-pack together reversibly at this interface is plausibly responsible for the higher levels of conductivity on the cooling cycle with either C16H33OH or C12H25OC6H5 components.
 | ||
| Fig. 3 Temperature dependence of conductivities of bulk systems. (——□) pure C16O5, (——△) complex C16O5∶LiClO4 (1∶1), (——●) C16O5∶C16H33OH∶LiClO4 (1∶1∶1), (——○) C16O5∶C12H25OPh∶LiClO4 (1∶1∶1). Arrows indicate the direction of heating. Dashed line: (EO)8∶Li(CF3SO2)2N. | ||
Langmuir–Blodgett films of the polymers 1 and the RX components are readily deposited as mixtures from an aqueous subphase as mono- or multi layers.9–13 However, the discovery that the salt-containing two- or three-component complexes with hydrophilic cations Na+ and Li+ could be deposited onto electrode surfaces either by adsorption of the salt from solution in the aqueous subphase or as a ‘pre-prepared complex' directly from chloroform solution was surprising. The presence of the salt as a complex with the organic components was indicated by the pressure–area isotherms12,13 and confirmed by grazing incidence FTIR12,13 and the electrical conductivity9–13 of the films collected on solid substrates.
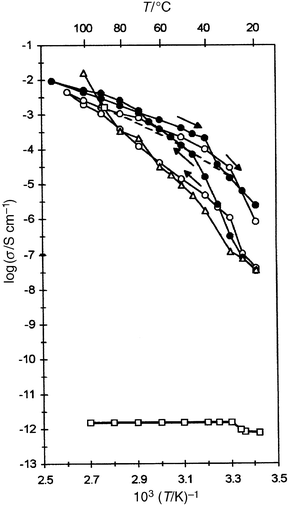
Fig. 4 shows pressure–area isotherms for C16O5, the two-component complex C16O5∶NaCF3SO3 formed both by taking up the salt from the aqueous subphase (Fig. 4b) and by depositing the ‘pre-prepared' (1∶1) complex onto the subphase (Fig. 4c). Fig. 4d shows the isotherm from deposition of the ‘pre-prepared' three-component system C16O5∶C20H41NH2∶NaCF3SO3 (1∶1∶1). The areas per monomer unit at a surface pressure of 25 mN m−1 increase as might be expected from 39 Å2 for C16O5 to ca. 70 Å2 for the two-component salt-containing systems and ca. 90 Å2 for the three-component complex. These results clearly indicate that salt has been taken up by the polymer from the subphase but also show, perhaps surprisingly, that a similar salt content is present by the ‘preprepared' procedure. The increase in area of 20 Å2 owing to C20H41NH2 is in line with expectation for the cross-sectional area of a n-alkyl chain. Also shown in Fig. 4e is the isotherm for pre-prepared C16O5∶C16H33OH∶ LiClO4 (1∶1∶1) which also has an area of ca. 90 Å2. Both three-component systems might be expected to have similar areas per repeat unit but such close correspondence between the two systems is perhaps coincidental. Different salt hydration energies might be expected to give rise to different equilibria between the aqueous subphase and the polyether helical environment unless the full equimolar proportion of salt has been retained in both cases.
 | ||
| Fig. 4 Surface pressure vs. area isotherms of Langmuir monolayers. (a) C16O5, (b) C16O5 in NaCF3SO3 containing subphase, (c) pre-prepared complex C16O5∶NaCF3SO3 (1∶1), (d) pre-prepared complex C16O5∶C20H41NH2∶NaCF3SO3 (1∶1∶1), (e) pre-prepared complex C16O5∶C16H33OH∶LiClO4 (1∶1∶1). | ||
The electrical conductivities of LB films of the above two- and three-component systems, are shown in Fig. 5 plotted as log10σversus 1/T. These measurements have been made using impedance spectroscopy with two vacuum-deposited electrodes above and below the LB films and so correspond to the conductivities through the films in the direction of a (see Fig. 2). Also shown are the conductivities of salt-free LB films of C16O5 measured in the same direction using an electrometer. Although the conductivities of the salt-containing LB films are lower than the corresponding bulk values shown in Fig. 3, it is clear that the addition of the various salts has effected an increase of 4–5 orders of magnitude over the salt-free LB film at temperatures between ambient and 80–90°C. However, most significantly approximate temperature-independent conductivity over this range has been observed in all the LB films of two- and three-component systems with C16O5 so far investigated.
 | ||
| Fig. 5 Temperature dependence of conductivities of multilayer LB films (18–20 layers). (——■) C16O5, (——●) pre-prepared C16O5∶C16H33OH∶LiClO4 (1∶1∶1), (——△) LB film of C16O5∶NaCF3SO3 from salt containing subphase, (——○) C16O5∶C20H41NH2∶ NaCF3SO3 (1∶1∶1) from pre-prepared complex. Arrows indicate the direction of heating. | ||
The lower conductivities may perhaps be explained in part by lower salt concentrations in the LB films than in the bulk films. However, since the pressure–area isotherms suggest that the LB films were deposited onto the electrode substrates with n-alkyl side-chains oriented normal to the electrodes (or approximately so) it might be supposed that the conduction in this direction would require ‘tunneling' through the ionophobic lamellae. This might be expected to give conductivities closer to those of the salt-free systems perhaps involving a more thermally activated process than is suggested by the data for the salt complexed systems in Fig. 5. A minority of experiments have been observed to give such low conductivity; but in most cases and with a variety of two- and three-component systems involving C16O5 the higher conductivity, low activation energy behaviour shown in Fig. 5 has been observed. Low activation energies might be expected to arise from conduction in the other directions, b or c (see Fig. 2), where virtually unimpeded pathways are possible. Alternatively, the data may arise from an experimental artifact to which the salt-containing systems, but not the salt-free ones, are susceptible. For the moment we refer to the low conductivity, thermally activated process in salt-containing systems as ‘Type 1' behaviour and the higher conductivity, low activation process as ‘Type 2'. These different types of results are considered in further detail in light of new experimental data in this paper.
With further temperature increase a decline in conductivity would seem to indicate a breakdown in the integrity of the films. However, on cooling the conductivities recover significantly before resuming the temperature-independent conduction at a lower level than initially as shown in Fig. 5 for the C16O5∶C16H33OH∶LiClO4 (1∶1∶1) system.
In this paper we compare the structure and conductivity of LB films and bulk samples of the complex of C16O5 and cetyl alcohol with the salt lithium bis(trifluoromethane)disulfonimide, (CF3SO2)2N− Li+, which has received considerable attention21,22 as a constituent of conventional amorphous polymer electrolytes for applications such as lithium batteries. LB films of the low-dimensional systems offer the possibility of measuring the anisotropy of ionic conduction along the molecular channels (b and c directions) and across the hydrocarbon layers (a direction). We have attempted to measure these quantities for the first time and the results are reported below. We also report new low-angle X-ray scattering results on a low-dimensional system and conductivity data on an LB film of a complex of 1 having R = octadecyl side chains, C18O5∶LiClO4. In addition we present, for the first time, a molecular dynamics model of a C16O5∶lithium salt complex (Fig. 1) and report details of the lithium ion coordination.
Experimental
The polymers 1, poly[2,5,8,14-pentaoxapentadecamethylene(5-hexadecyloxy-1,3-phenylene)], C16O5, and the analogous octadecyl derivative, C18O5, were prepared as described previously.3,5 Cetyl alcohol and LiClO4 were obtained from Aldrich Chemicals and used as supplied. Li(CF3SO2)2N was obtained from Fluka and used as supplied. Complexes with LiClO4 or Li(CF3SO2)2N were prepared in acetone–chloroform mixtures. After removal of the solvent the complexes were held at 60°C under vacuum for 1 day to ensure distribution of the salts throughout the system.Langmuir films were deposited from chloroform solutions of equimolar two- or three-component complexes of concentration ≅0.5 mg ml−1 onto ultra purified water at pH 6.0 (the ‘pre-prepared' procedure). Surface pressure π was continuously monitored using a Wilhelmy plate and electrobalance arrangement. LB films were transferred (Y-type) onto glass slides having appropriate metallised electrode patterns at π = 20 mN m−1 at a vertical dipping rate of 4–8 m min−1. Films of between 18 and 31 molecular layers were prepared. For measurements in the bc plane (see Fig. 2) a pattern of interdigitated gold electrodes (100 nm thickness) as shown in Fig. 6a were used. There were 21 pairs of electrodes in total with electrode length 1.5 cm and electrode separation 130 μm. The films were deposited by dipping the slides in each direction ‘electrode parallel' and ‘electrode perpendicular' in the event that molecular orientation during the transfer process could reveal some anisotropy of conduction between b and c molecular directions. For measurements in the a direction patterns of upper and lower aluminum electrodes (50 nm thickness) as shown in Fig. 6b were used. After LB film deposition onto the lower electrode the upper electrode was deposited by slow evaporation of aluminum to avoid excessive heating of the film (50 Å at 1 Å s−1, 150 Å at 3–5 Å s−1 and 350 Å at 8 Å s−1). The total area of overlap of upper and lower electrodes was 0.75 cm2. Conductivity measurements were performed under vacuum with the slides mounted on a thermostatically controlled heating block in a sealed chamber. For the measurement of bulk conductivity of C16O5∶C16H33OH∶Li(CF3SO2)2N (1∶1∶1) the sample was enclosed between ITO electrodes with a 100 μm spacer. Impedance spectra were measured over the frequency range 0.01–64 kHz with amplitudes of 1 V for the bulk system, 10 V for the ‘in-plane' (bc directions) LB films with interdigitated electrodes and 10–100 mV for the ‘normal-to-plane' (a direction) LB films.
 | ||
| Fig. 6 Schematic representation of electrodes for conductivity measurements of LB films. (a) Electrode arrangements for ‘in-plane' measurements. The experimental grids used 21 pairs of gold electrodes 100 nm in thickness. Electrodes were 1.5 cm in length with 130 μm separation. (b) Electrode arrangement for ‘normal-to-plane' measurements. Electrodes were of 50 nm aluminium and the total area of overlap between upper and lower electrodes was 0.75 cm2. | ||
Small-angle X-ray scattering was measured on equipment specially optimised for LB work at the Université du Maino, France, using a Philips PW 1130 X-ray generator, graphite monochromatised Cu-Kα (λ = 1.54 Å) radiation and a MAR Research image plate area detector.
Molecular dynamics modelling was performed with Cerius2 molecular dynamics software. The model was done with ‘annealing cycles' between room temperature and 600 K.
Results and discussion
Molecular dynamics modelling of C16O5: lithium salt complex
Fig. 1 shows a ‘snapshot' of 100 such annealing cycles at ambient temperature. In this ‘snapshot' it is apparent that the lithium ion is in different locations within the ‘opposing' polyether helices. When the full sequence of annealing cycles is run it is evident that the lithium ‘rattles' between different locations within the helices. It is also evident in Fig. 1 that some oxygens are not directed ‘in' towards the helical axis. Inspection of the other ‘snapshots' in the sequence reveals that in no conformational sequence of the polyether segment are all of the oxygens directed ‘in' to the cation. When compared with the ‘ideal' conformations of t t g about C–O–C–C sequences in PEO–alkali cation complexes14–18 it is evident that this should have a relatively destabilising effect on the lithium ion in C16O5. As discussed in the Introduction such destabilisation should be beneficial in promoting cation mobility within the helices. This point can be emphasised by inspecting the bond rotational angles in Fig. 1 (trans (t) = 180 ± 60°, gauche (g) = 60 ± 60°, minus gauche ( ) = −60 ± 60° and cis = 0°).
) = −60 ± 60° and cis = 0°).
In contrast to the ‘ideal' sequences of the PEO complexes there are five C–O bonds in gauche states and four C–C bonds in trans states in the C16O5 complexes. The sequence of snapshots reveals that the hexadecyl side chains are generally aligned in parallel interdigitated array although, as in Fig. 1, departures from the lowest energy all-trans conformations occur at ambient temperatures within the overall hexagonal lattice.
Low-angle X-ray scattering of C16O5∶LiClO4. Langmuir–Blodgett film
Fig. 7 shows an X-ray scattering trace of an LB film of 18 molecular layers of C16O5∶LiClO4. Four orders of reflection are apparent confirming that these complexes form LB films having a highly organised laminar structure. From the third-order peak at 2θ = 7.1 Å and the fourth-order peak at 2θ = 9.6 Å, values for the long spacing of 37.4 Å and 36.9 Å, respectively, may be calculated. Comparing with measured values for crystalline bulk samples at ambient temperatures for pure C16O5 and for C16O5∶LiClO4 (1∶1) of 35.6 Å and 39.2 Å respectively, it is apparent that the long spacing for the LB film lies between these two. In light of the molecular models shown in Fig. 1 and 2 this observation is apparently consistent with Y-type interdigitated deposition of molecular layers. However, the intermediate value of the long spacing presumably indicates less than equimolar retention of salt in the deposited LB film. | ||
| Fig. 7 Reflectivity vs. scattering angle 2θ for LB film of ‘pre-prepared' complex C16O5∶LiClO4 (1∶1). | ||
Conductivities of bulk two- and three-component complexes with Li(CF3SO2)2N
It is immediately apparent from hot-stage polarised light microscopy on the bulk films that two- and three-component systems with (CF3SO2)2N− anions do not form extensive smectic structures in the crystalline phase nor do they form a liquid-crystal phase at temperatures above side chain melting (34–37°C). Whereas the systems described in Fig. 3 are highly birefringent with well defined ‘broken fan' textures indicative of smectic A morphology3–8,19,20 the complexes with Li(CF3-SO2)2N have ‘granular' textures below ca. 33°C becoming apparently isotropic after side chain melting. A plausible explanation for these observations is apparent from Fig. 2 and concerns the relation between the stability of an extensive two-dimensional structure and the anion size. Anions such as ClO4−, BF4−, CF3SO3− and Br− are small enough (≤
ca. 4.5 Å diameter) to occupy adjacent sites alongside each Li+ encapsulated along the helix in an equimolar complex. In the crystalline state of the side chains the inter-lithium distances are determined by the dimensions of the hexagonal lattice of the alkane chains which are ca. 4.2 Å apart along the helix and ca. 7.3 Å between adjacent helices. These distances may increase in the liquid crystalline states. However, given the added constraints of associating with the lithium ions in the C16O5 helices it seems likely that equimolar complexes with the large (CF3SO2)2N− anion are unable to sustain an extensive two-dimensional surface. The complex may therefore adopt a ‘micellar' arrangement of the ionophobic and ionophilic components of the molecular complex which allows maximum surface area for the cation–anion interactions. Since no decline in the ionic conductivity of the liquid-crystal forming systems has been observed at the liquid crystal-to-isotropic transitions (80–110°C for most three-component systems) it seems likely that the highest conductivities observed in Fig. 3 may correspond to micellar textures creating ‘pseudo three-dimensional' conduction pathways between the micelles. If such structures were well organised ions would be confined to surface layers of ionophilic helical material in which trapping within ionophobic pockets would be avoided. Nonetheless, our experiments with heat treatment and orientation of the complexes with the smaller anions indicate6,7 that extensive pre-organisation of a liquid crystal phase is a useful precursor to the most conductive textures.Fig. 8 shows log10σversusT−1 plots for bulk conductivities of C16O5∶Li(CF3SO2)2N (1∶1), C16O5∶C18H38∶Li(CF3-SO2)2N (1∶1∶1) and C16O5∶C16H33OH∶Li(CF3SO2)2N (1∶1∶1). Notwithstanding the absence of a liquid crystal phase the levels of conductivity above side chain melting are encouraging. The conductivities of the two-component system and of C16O5∶C18H38∶Li(CF3SO2)2N (1∶1∶1) above ca. 30°C compare very favorably with that reported by Sylla and coworkers23 for PEO–Li(CF3SO2)2N. The data for C16O5∶C16H33OH∶Li(CF3SO2)2N (1∶1∶1) exceed those of the PEO system over this range and are similar to the levels achieved in the cooling runs of the three-component systems in Fig. 3. It seems possible that the –OH or the phenoxy groups O–C6H5 (see Fig. 3) may be more effective than the alkane C18H38 as surfactants in stabilising micellar surfaces and maximising the areas of conducting surfaces. The ‘step' in conductivity of the C16O5∶Li(CF3SO2)2N (1∶1) system at 80–100°C (reproduced in repeated runs of this system) is an intriguing feature which may involve a structural rearrangement arising from the spatial requirements of the anion discussed above. It seems significant that the step is absent in the three-component systems which have greater surface areas per ion pair.
 | ||
| Fig. 8 (a) Temperature dependence of conductivities of bulk systems and in-plane measurements of multilayer LB films (see Fig. 6a). Bulk systems: (——□) C16O5∶LiTFSI (1∶1), (——●) C16O5∶C18H38∶ LiTFSI (1∶1∶1), (——○) C16O5∶C16H33OH∶LiTFSI (1∶1∶1); C16O5∶C16H33OH∶LiTFSI (1∶1∶1) LB films (31 layers): (——■) dipping direction parallel to electrodes (see Fig. 6a), (——△) dipping direction perpendicular to electrodes. Dashed line: (EO)8∶ Li(CF3SO2)2N. Arrows indicate the direction of heating. (b) Typical impedance plots for in-plane measurements of LB films. (□) 30°C, (■) 60°C, (○) 90°C, (△) 120°C. | ||
Langmuir–Blodgett films of C16O5∶C16H33OH∶Li(CF3SO2)2N
Isotherms of surface pressure versus area per polymer repeating unit for C16O5 and a pre-prepared complex of C16O5∶C16H33OH∶Li(CF3SO2)2N (1∶1∶1) are shown in Fig. 9. The difference between each isotherm in area per repeating unit at 25 mN m−1 is ca. 20 Å2. As in Fig. 4, this additional area is consistent with that required by an n-alkyl molecule normal to the surface of the subphase. However, Fig. 4 also indicates that the salt in the complex contributes 30–35 Å2 to the area. The location of the salt in the surface monolayer is a matter for speculation and is likely to depend on the nature of the salt. The results of Fig. 4 suggest that both anions CF3SO3− and ClO4− (probably hydrated) may contribute to the area by lying alongside the helix. An encapsulated cation may also dilate the helix as well as promoting hydration with a structure atypical of the liquid water. More weakly associated anions may lie beneath the helix in the aqueous subphase and may thus contribute less to surface area. Nonetheless, the total area in Fig. 9 of only ca. 60 Å2 at 25 mN m−1 suggests, by comparison with Fig. 4, that little of the Li(CF3SO2)2N salt may be associated. This may perhaps find an explanation, at least in part, in spatial arguments proposed for the failure of (CF3SO2)2N− to form a liquid crystal phase as discussed above, i.e. the anion is too large to be accommodated alongside lithium ions occupying adjacent sites in extensive two-dimensional monolayers of the complexes. Some preliminary examination by grazing-incidence FTIR of the LB multilayer films deposited from the monolayers also indicates low salt concentrations since the characteristic anion bands, such as the SO2 stretch at 1300–1415 cm−1 appear to be weak. | ||
| Fig. 9 Surface pressure vs. area isotherms of Langmuir monolayers. (a) C16O5, (b) C16O5∶C16H33OH∶LiTFSI (1∶1∶1). | ||
The temperature dependence of the ‘in-plane' ionic conductivity of the LB film of the C16O5∶C16H33OH∶Li(CF3-SO2)2N system deposited onto the interdigitated electrode grid (Fig. 6a) is shown in the lower section of Fig. 8a. The ‘three-dimensional conductivities' were calculated by assuming a long spacing of 40 Å per pair of molecular layers. The results for the different depositions both parallel and perpendicular to the electrodes are very similar suggesting that different molecular orientations in the bc plane (see Fig. 2) are either absent or do not bring about anisotropy in conduction. The impedance plots of these thin films over frequencies from 0.1 Hz to 64 kHz were approximately semicircular as shown in Fig. 8b. Some evidence for a second semicircle is present at lower temperatures but it was assumed that the Z′ values at the non-zero minimum represented the ‘in-plane' resistances of the films.
The temperature dependence of the ‘normal-to-plane' conductivities of the LB films of C16O5∶C16H33OH∶Li(CF3-SO2)2N are given in Fig. 10a as log10σversusT−1 (where σ is the three-dimensional conductivity calculated as for the in-plane data). As described in the Introduction, in this orientation of the electrodes two kinds of behaviour have been observed in all films so far investigated and including the C16O5∶C16H33OH∶Li(CF3SO2)2N system. ‘Type 1' conduction, which is characterised by low conductivity of the order of 10−12 S cm−1 increasing with temperature, is the ‘expected behaviour' for measurements normal to the plane of a multilayer constructed from alternating ion-containing surfaces and insulating hydrocarbon layers which are parallel to the electrodes. Such behaviour should be similar to that of the salt-free films shown by the electrometer measurements in Fig. 5. In the present work the Type 1 conductivity was too low for precise resistance measurements using the FRA/ECI. However, by assuming semicircular impedance plots the resistances at various temperatures were estimated from impedance data over 64 kHz to 0.1 Hz shown in Fig. 10b. Type 1 conductivity of the C16O5∶C16H33OH∶Li(CF3SO2)2N film is shown in the lower points labeled ‘i' in Fig. 10a.
 | ||
| Fig. 10 (a) Temperature dependence of normal-to-plane conductivity of C16O5∶C16H33OH∶LiTFSI (1∶1∶1) multilayer (31 layers) LB films (see Fig. 6a). Arrows indicate the direction of heating. (i) estimated conductivities from incomplete semicircles (see Fig. 10b), (ii) sample opened to atmosphere; vacuum reapplied on reheating above 20°C. (b) Typical impedance plots of LB film, ‘type 1' behaviour. (c ) Typical impedance plots of LB film, ‘type 2' behaviour. | ||
However, in most samples, after the first heating to ca. 50°C a sharp increase in conductivity to 10−7–10−6 S cm−1 is observed as shown in Fig. 10a. This level of conductivity, which is also shown for LB films of other systems in Fig. 5, is retained in subsequent heating and cooling over the temperature range between ambient and ca. 80–90°C. On cooling C16O5 complexes below ca. 20°C the conductivity decreases. Although the effect is small in this sample it may be attributed to crystallisation of the side chains as observed in bulk samples at higher temperatures. It provides some evidence that this ‘Type 2' behaviour is characteristic of the LB film rather than being an experimental artifact possibly arising from a change in the electrode arrangement. In the present experiment the vacuum was removed on cooling to 10°C. As shown at ‘ii' in Fig. 10a this brought about the expected rise in conductivity by a factor of 3 as moisture entered the sample. On reheating under vacuum the conductivity returned to the former level at ca. 10−7 S cm−1 providing further evidence that Type 2 behaviour reflects a change in the conductivity of the film. As Fig. 10c shows the impedance plots for Type 2 behaviour are good semicircles giving unambiguous intercepts on the Z′ axis. The high frequency points for each temperature extrapolate to between 5 and 10 Ω which is the estimated resistance of the aluminum electrodes. The level of Type 2 conductivity for this system lies below that of the results (heating runs) reported in Fig. 5. This is consistent with a lower concentration of salt in the present system as suggested by the pressure–area isotherms.
With heating above ca. 90°C the conductivity departs from the apparent temperature independent behaviour. In other systems such as C16O5∶C16H33OH∶LiClO4 shown in Fig. 5 this effect is more dramatic than in the present system. In that system the conductivity plunges four orders of magnitude but on recooling it partially recovers and resumes temperature-independence. This phenomenon presumably indicates some irreversible destruction of the integrity of the film although no evidence can be discerned from optical microscopic examination of the upper aluminum electrodes.
Fig. 11 shows apparent Type 2 behaviour in an LB film of C18O5∶LiClO4. The longer octadecyl side chain recrystallises more readily than the hexadecyl one and this presumably accounts for a more pronounced fall in the conductivity on cooling below 20°C. The temperature independent region is less extensive than in C16O5 films.
Whilst we remain alert to the possibility that Type 2 behaviour is an experimental artifact not characteristic of the LB film conductivity we assume for the moment that a transition in the morphological structure of the complex occurs at temperatures ≥ca. 50°C during the first heating run of the majority of samples. We are as yet unable to identify an experimental factor which selects Type 2 behaviour in the majority of experiments and the occasional failure to transform from Type 1. However, we tentatively propose that the transition to Type 2 behaviour represents a reorientation of the conductive ion-containing surfaces within the films so that they lie normal to the electrodes. This should give rise to a considerable increase in conductivity and unimpeded pathways for at least the anions could account for the negligible activation energy.
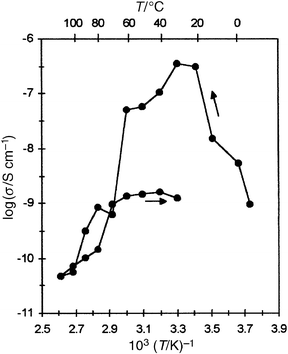 | ||
| Fig. 11 Temperature dependence of normal-to-plane conductivity of a multilayer C18O5∶LiClO4 (1∶1) LB film (20 layers). Arrows indicate the direction of heating. | ||
Since no transition in the ‘in-plane' conductivity is apparent in Fig. 8a it seems possible that the transition is brought about by the presence of the upper electrode in the ‘normal-to-plane' measurements. The driving force for the transition may be associated with a change to a lower energy of interaction at the interface between the electrodes and the polymer. Alternatively, a change in orientation could arise from anisotropy in the thermal expansion of the low-dimensional system confined within a fixed space between the electrodes. Over the temperature range 40–60°C two transitions are observed in the conductivities of ‘small anion' systems corresponding to side chain melting in the region of 40°C followed by a second in the region of 60°C which we attribute to an increase in the interlamellar space occupied by the anions (see Fig. 3). Even before the second transition in the liquid crystal state we have observed such an increase in a number of systems such as C16O5∶LiClO4 (1∶1) which increases from 39.2 Å at 25°C to 43.7 Å at 55°C. In the present ‘large anion' complex the two transitions apparently coincide at side chain melting so that larger than 10% expansion in this direction could be anticipated. If a smaller thermal expansion occurs in one of the remaining dimensions the temperature increase may prompt a reorientation to allow the larger expansion in the plane of the film. It might be supposed that the most facile reorientation of the high polymer chains would involve a rotation about the extended helical axis leaving both the helices and the alkyl side chains lying parallel to the electrodes. Such a rotation would cause the interhelical c direction (see Fig. 2) to orient perpendicular to the electrodes. If our earlier conjecture that conductivity in the b direction may exceed that in the c direction is correct, this interpretation would be consistent with the LB data of Fig. 8a which indicate that the ‘in-plane' (bc) conductivity can exceed the Type 2 conductivity above ca. 60°C. The significant temperature dependence of these bc data may reflect the low concentrations of salt creating heterogeneities in structure over the large inter-electrode distances. Further experiments to test these hypotheses are in progress.
Conclusions
i) We have prepared LB multilayers (up to 31 molecular layers) as well as ‘bulk' (100 μm thickness) films of low dimensional polymer electrolytes C16O5∶C16H33OH∶Li(CF3SO2)2N. Low-angle X-ray diffraction measurements confirm the laminar structure of these films and the measured long spacings of the LB films (ca. 37 Å) also support earlier findings from FTIR spectra that the salt loadings are lower than the bulk equimolar formulations.ii) The ‘bulk' films gave conductivities generally higher than O–Li(CF3SO2)2N over the temperature range between alkyl side-chain melting at 40°C (σ = 6 × 10−4 S cm−1) and 100°C (σ = 8 × 10−3 S cm−1).
iii) The in-plane conductivity measurements of the LB films, measured for the first time with interdigitated electrodes, gave linear Arrhenius plots with activation energy 79.4 kJ mol−1 between ambient temperature and 110°C (σ
∼ 10−5 S cm−1).
iv) The normal-to-plane measurements of the LB films confirmed previous observations on several different low-dimensional LB systems of this kind in exhibiting temperature independent conductivity over the range 10 to 90°C at a conductivity level of 10−6 to 10−7 S cm−1. Given the low salt content of these samples such behaviour was unexpected if the helical planes within which the ions are mobile remained parallel to the electrodes following deposition of the LB films. However, in this work we have established that very low temperature-dependent conductivities are indeed observed (ca. 10−12 S cm−1) during the first heating cycle below ca. 50°C but at a sharp transition at this temperature an apparently irreversible transformation to the higher conductivity, temperature-independent behaviour occurs.
v) We conclude that a structural rearrangement of the films occurs during the first heating cycle and propose that the molecular layers rotate so as to reorient the planes of high ion mobility normal to the electrodes. Such a rearrangement may arise in response to the anisotropy in thermal expansion in the confined space between upper and lower electrodes.
vi) That such structural rearrangements are possible in the LB systems has encouraging implications for similar reorientations and the optimisation of conductivity at ambient temperatures in bulk films.
Acknowledgements
We acknowledge a grant from EPSRC for support in this area.References
- C. A. Angell, C. Liu and E. Sanchez, Nature, 1993, 362, 137 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456 CrossRef CAS.
- F. B. Dias, J. P. Voss, S. V. Batty, P. V. Wright and G. Ungar, Macromol. Rapid Commun., 1994, 15, 961 CrossRef CAS.
- F. B. Dias, S. V. Batty, J. P. Voss, G. Ungar and P. V. Wright, Solid State Ionics, 1996, 85, 43 Search PubMed.
- F. B. Dias, S. V. Batty, G. Ungar, J. P. Voss and P. V. Wright, J. Chem. Soc., Faraday Trans., 1996, 92, 2599 RSC.
- F. B. Dias, S. V. Batty, A. Gupta, G. Ungar, J. P. Voss and P. V. Wright, Electrochim. Acta, 1998, 43, 1217 CrossRef CAS.
- P. V. Wright, Y. Zheng, D. Bhatt, T. Richardson and G. Ungar, Polym. Int., 1998, 47, 34 CrossRef CAS.
- A. Tajbakhsh, F. Dias, Y. Zheng and P. V. Wright, Mol. Cryst. Liq. Cryst., 1998, 323, 69 Search PubMed.
- S. V. Batty, T. Richardson, F. B. Dias, J. P. Voss, P. V. Wright and G. Ungar, Thin Solid Films, 1996, 284–5, 530 CrossRef.
- Y. Zheng, D. Bhatt, F. B. Dias, S. V. Batty, A. Tajbakhsh, T. Richardson, P. V. Wright and G. Ungar, Supramolecular Science, 1997, 4, 525 Search PubMed.
- Y. Zheng, F. B. Dias, P. V. Wright, G. Ungar, D. Bhatt, S. V. Batty and T. Richardson, Electrochim. Acta, 1998, 43, 1633 CrossRef CAS.
- Y. Zheng, D. Bhatt, F. Davis, T. Richardson and P. V. Wright, Thin Solid Films, 1998, 327–9, 473 CrossRef.
- D. Bhatt, Y. Zheng, T. Richardson, P. V. Wright and F. Davis, Thin Solid Films, 1998, 327–9, 364 CrossRef.
- Y. Chatani and S. Okamura, Polymer, 1987, 28, 1815 CrossRef CAS.
- Y. Chatani, I. Y. Fuji, T. Takayanagi and A. Honma, Polymer, 1990, 31, 2238 CrossRef CAS.
- P. Lightfoot, M. Mehta and P. G. Bruce, J. Mater. Chem., 1992, 2, 379 RSC.
- P. Lightfoot, M. Mehta and P. G. Bruce, Science, 1993, 262, 883 CrossRef CAS.
- P. Lightfoot, J. L. Nowinski and P. G. Bruce, J. Am. Chem. Soc., 1994, 116, 7469 CrossRef CAS.
- F. B. Dias, PhD thesis, University of Sheffield, 1996. Search PubMed.
- Y. Zheng, PhD thesis, University of Sheffield, 1998. Search PubMed.
- D. Benrabah, D. Baril, J. Y. Sanchez and M. Armand, J. Chem. Soc., Faraday Trans., 1993, 89, 355 RSC.
- F. Alloin, J. Y. Sanchez and M. Armand, Solid State Ionics, 1993, 60, 3 Search PubMed.
- S. Sylla, J. Y. Sanchez and M. B. Armand, Electrochim. Acta, 1992, 37, 1699 CrossRef CAS.
Footnote |
| † Basis of a presentation given at Materials Chemistry Discussion No. 2, 13–15 September 1999, University of Nottingham, UK. |
| This journal is © The Royal Society of Chemistry 2000 |