Diode-like electron transfer across nanostructured films containing a redox ligand†
David I. Gittins, Donald Bethell, Richard J. Nichols and David J. Schiffrin*
Department of Chemistry, University of Liverpool, Liverpool, UK L69 7ZD
First published on UnassignedUnassigned22nd December 1999
Abstract
Novel redox active dithiols containing the viologen (4,4′-bipyridinium group) have been synthesised. Monolayers and multilayers consisting of alternating layers of the dithiol and gold nanoparticles have been self-assembled on a gold substrate and visualised using SPM. Such materials have been investigated for their electroactivity and their ability to mediate electron transfer to an external redox couple, [Ru(NH3)6]3+/2+, in aqueous solution. Viologen moieties in these multilayers are addressable electrochemically provided they are within 6–8 layers of the external surface and it is shown that electron transfer through the deeper layers is very facile as a consequence of the presence of partially filled viologen π-orbitals. When the viologen containing dithiol is present in the outer layer, a diode effect is observed; electron transfer from the multilayer to the external couple takes place, but the reverse electron transport is inhibited.
Introduction
Gold nanoparticles, discrete gold clusters of diameter 1–100 nm, first studied by Faraday,1 have been recently re-discovered as versatile objects with potential for use in the miniaturisation of electronic devices, such as the single electron transistor.2,3 A recent observation by Brust et al. of a synthetic method to reduce gold salts in toluene in the presence of an alkanethiol resulted in stable nanoparticles in which the thiol formed a protective shell.4 Particles synthesised by this method maintain their integrity when the solvent is removed and they behave similarly to chemical compounds. Unlike the earlier hydrosols this synthetic procedure, combined with self-assembling methods, have already produced several important advances in the emerging field of nanotechnology. The study of derivatised particles of size between that of atoms and bulk solids has led to the observation of single electron transfer and the self assembly of nanostructured materials.5–19 Recent reviews by Feldheim,2 Schmid,3 and Antonietti and Goltner20 illustrate in greater detail the role of chemistry in this new science.Electron transfer across arrays of gold nanoparticles linked or separated by alkane thiols has been demonstrated,7 but the influence of the electronic structure of the linker on electronic properties is not known at present.17,19 We report here the assembly of an electroactive nanostructured material that preserves the integrity and dimensions of the gold nanoparticles used in its construction. We also describe experiments that characterise some of its electrical and other physical properties and demonstrate the addressability of the electroactive groups in the film by simple electrochemical methods. The redox linker employed was a viologen dithiol (1). Viologens (1,1′-dialkyl-4,4′- bipyridinium salts) have been previously widely studied for their reversible redox activity and stability and were employed for this reason as linkers in the present work.21
Experimental
Synthesis
Except where stated all chemicals were used as received. The organic solvents were of analytical grade or purified by standard laboratory techniques; water was purified using a Millipore MQ system. All reactions were stirred magnetically and monitored by thin layer chromatography (TLC), using aluminium-backed silica gel plates coated with Merck 60F-254 silica gel. Flash column chromatography was performed using Merck 60 silica gel (40–60 μm). Nuclear magnetic resonance 1H spectra were recorded on a Varian 300 Gemini 2000 spectrometer. All chemical shifts (δ) are quoted in parts per million (ppm) and are reported relative to an internal tetramethylsilane (TMS, δ 0.00) standard. Infrared spectra were recorded on a Perkin Elmer 881 infrared spectrophotometer. FAB+ mass spectra were recorded on a VG7070E spectrometer (Micromass), GC-MS spectra were recorded on a TRIO1000 spectrometer (Fisons). Microanalyses were performed using a Carlo Erba elemental analyser in the Department of Chemistry Microanalytical Laboratory at Liverpool.1-Acetylthio-10-bromodecane
Potassium thioacetate (10.68 mmol, 1.22 g) and 1,10-dibromodecane (10.68 mmol, 3.21 g) were refluxed in dry tetrahydrofuran (200 mL) overnight. The cooled solution was then filtered to remove potassium bromide. GC-MS analysis of the product mixture gave 20% 1,10-dibromodecane, 53% desired product and 27% 1,10-bis(acetylthio)decane. The starting material was removed on a silica column with petroleum ether (bp 40–60![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) °C) [monitored on TLC 90:10 hexane–ethyl acetate, anisaldehyde visualisation, Rf 0.36, 0.51, 0.64 (1,10-dibromodecane, 1-acetylthio-10-bromodecane, 1,10-bis(acetylthio)decane respectively)]. The product was eluted with increasing concentrations of ethyl acetate, the solvent removed by rotary evaporation and dried in vacuo to give a golden waxy solid. Total mass of product 4.06 g. Analysis by GC-MS gave 1,10-dibromodecane 1%, 1-acetylthio-10-bromodecane 61%, 1,10-bis(acetylthio)decane 39%. Desired product yield 2.47 g (47%). IR (KBr plates, neat, cm−1), ν(CO) 1695.
°C) [monitored on TLC 90:10 hexane–ethyl acetate, anisaldehyde visualisation, Rf 0.36, 0.51, 0.64 (1,10-dibromodecane, 1-acetylthio-10-bromodecane, 1,10-bis(acetylthio)decane respectively)]. The product was eluted with increasing concentrations of ethyl acetate, the solvent removed by rotary evaporation and dried in vacuo to give a golden waxy solid. Total mass of product 4.06 g. Analysis by GC-MS gave 1,10-dibromodecane 1%, 1-acetylthio-10-bromodecane 61%, 1,10-bis(acetylthio)decane 39%. Desired product yield 2.47 g (47%). IR (KBr plates, neat, cm−1), ν(CO) 1695.N,N′-Bis(10-acetylthiodecyl)-4,4′-bipyridinium dibromide
The purified product mixture taken from the previous step containing 1-acetylthio-10-bromodecane (4.1 mmol, 1.22 g) was melted with 4,4′-bipyridyl (1.6 mmol, 0.25 g) and maintained at 80°C for four days. TLC (ethanol, UV visualisation) indicated that all the 4,4′-bipyridyl had reacted. Methanol (50 mL) was added to give a golden oil. Yellow crystals precipitated on addition of diethyl ether (250 mL) which were collected and washed with diethyl ether (250 mL). Yield 1.18 g (97%). Found C 54.5, H 7.2, N 3.5, M+H+ 745.20810. C34H54N2S2O2Br2 requires C 54.7, H 7.3, N 3.8%, M+H+ 745.20717. 1H NMR(MeOD) δ 1.1–1.9 (br m, 32H, CH2 chain centre), 2.10 (br m, 4H, CH2CH2N+), 2.28 (s, 6H, CH2SAc), 2.85 (t, 4H, CH2SAc), 4.73 (p, 4H, CH2N+), 8.70 (d, 4H, 3,3′-H bipy), 9.31 (d, 4H, 2,2′-H bipy). m/z (FAB+) 586 [M− (Br)2]+. IR (KBr pellet, cm−1), ν(CO) 1692.N,N′-Bis(10-mercaptodecyl)-4,4′-bipyridinium dibromide (1)
N,N′-Bis(10-acetylthiodecyl)-4,4′-bipyridinium dibromide (0.71 mmol, 0.68 g) was dissolved in a minimum volume of dry degassed methanol at −78°C. The sample was purged with nitrogen during dropwise addition of excess acetyl chloride (~5 mL), the mixture was kept in dry ice for 5 minutes and allowed to warm to room temperature over a period of 3 h in a nitrogen atmosphere. All by-products were distilled off along with the solvent by rotary evaporation to leave the dithiol as a sticky clear golden fluid. Yield 0.62 g (100%). Found M+H+ 661.18523, C30H50N2S2Br2 requires 661.18604. 1H NMR(MeOD) δ 1.1–1.9 (br m, 32H, CH2 chain centre), 2.19 (br m, 4H, CH2CH2N+), 2.47 (m, 2H, CH2SH), 4.74 (p, 4H, CH2N+), 8.69 (d, 4H, 3,3′-H bipy), 9.39 (d, 4H, 2,2′-H bipy). m/z (FAB+) 502 [M− (Br)2]+. IR (KBr plates, neat, cm−1), ν(CO) not seen.The synthesis of underivatised gold nanoparticles was carried out as described previously.4,13 TEM analysis showed that the particles had an average diameter of 6 ±1 nm.
Sample preparation
Electrodes were derivatised by immersion in 1 mM methanol solutions of the viologen (1). To monitor the progress of monolayer formation, the electrode was removed periodically and rinsed first with methanol and then with water, followed by cyclic voltammetric measurement. A full monolayer was always formed after 18 hours of immersion as shown by the constant value of the peak currents. For the electrochemical examination of layered materials, the bulk gold electrode derivatised with (1) was subjected to a 3 hour immersion in a toluene solution of gold nanoparticles.13,17,22 Further layers were added by alternate dipping of the electrode in solutions of (1) and of gold nanoparticles in toluene. The type of structure expected by this method is schematically shown in Fig. 1. After each derivatisation step cyclic voltammograms were recorded to monitor the growth process. This gave different voltammetric responses depending on whether the surface was terminated by gold nanoparticles or by thiol. | ||
| Fig. 1 Schematic illustration of multilayer growth by alternate attachment of (1) and of gold nanoparticles. | ||
For spectroscopic examination, layered materials were grown on standard glass microscope slides (BDH); these were cleaned in fresh piranha solution (70:30 H2SO4–H2O2; CARE, these solutions must not be stored), rinsed with water and silanised with mercaptopropyl methyldimethoxysilane (Fluka) following the method of Goss et al.23 The thiol derivatised slides were then immersed in the toluene solution of gold nanoparticles for a minimum of 3 h followed by immersion in 1 mM methanol solution of the viologen for a minimum of 18 h. During transfer between the toluene and methanol solutions the slides were washed extensively with both solvents to avoid cross-contamination.
Cyclic voltammetry
Cyclic voltammetry experiments were carried out in a standard three electrode cell using an IBM PC controlled waveform generator and potentiostat (EcoChemie Pgstat20) to control the potential. The solutions were deaerated with high purity nitrogen for ten minutes before voltammograms were recorded. All potentials refer to the standard saturated calomel electrode (SCE). The buffered electrolyte used in all the electrochemical experiments was made by mixing 0.1 M solutions of potassium dihydrogen phosphate and dipotassium hydrogen phosphate (Fluka) until pH 7 was reached. Gold electrodes were prepared from 1 mm diameter gold wire (99.9985%, Johnson Matthey) sealed in soda glass and carefully melted to form a spheroid. Before use the electrodes were cleaned in fresh piranha solution and cycled between −0.5 V and 1.8 V at 0.5 V s−1 in 0.5 M H2SO4 until a typical voltammogram for clean polycrystalline gold was obtained. The electrode area was calculated from the sweep rate dependence of the voltammograms of 1 mM hexaammineruthenium(III) chloride using the Randles–Sevcik equation taking the diffusion coefficient as 7.5 × 10−6 cm2 s−1.24 Surface roughness, used for the coverage calculations, was measured by iodine adsorption.25Tapping mode atomic force microscopy (AFM)
Surface imaging was carried out using standard tapping mode tips in air on a NanoScope IIIa system (Digital Instruments). The gold substrates for the microscopy experiments were prepared by thermal evaporation under vacuum (Edwards 306A) of ~200 nm gold onto clean glass slides (onto which a ~2 nm chromium film had been previously deposited to assist adhesion). Before derivatisation samples were cleaned in piranha solution, rinsed, dried and flame annealed to produce large flat terraces with Au(111) structure.UV–VIS spectroscopy
Measurements were made with a HP8452 diode array spectrometer (Hewlett Packard); spectra were referenced to an underivatised glass slide.Results and discussion
Multilayer growth and analysis
The viologens were originally investigated electrochemically as redox indicators in biological studies and are the parent compounds of the most widely used herbicides, the ‘paraquat' group.21 More recently they have been used in biological and chemical systems as electron mediators, because of their electrochemically reversible behaviour and stability arising from low-lying empty π-orbitals. The viologens exist in three main oxidation states; dication (most stable), radical cation and diradical. They exhibit reversible voltammetry when reducing the dication to its radical form: bipm2+ + e− → bipm•+.Fig. 2 shows a typical cyclic voltammogram (CV) of the gold electrode bearing a monolayer of (1) and confirms that the electroactive species are adsorbed on the electrode surface (difference in peak potentials, ΔEp≈0 and peak current is proportional to the sweep rate [not shown]). This monolayer had a calculated surface coverage (Γ) of 4.1 × 10−10 mol cm−2, which compares well with a Γmax of 4.3 × 10−10 mol cm−2 assuming that each bipyridyl group is perpendicular to a perfect gold (111) surface.26 As the number of gold layers is increased, as illustrated in Fig. 1, the amount of electroactive viologen also increases. The dependence of the cathodic peak current density (jpc) on number of layers (Fig. 3a) provides evidence of this.
 | ||
| Fig. 2 Cyclic voltammogram of the first monolayer of (1) at saturation coverage (Γsat = 4.1 × 10−10 mol cm−2). 0.1 M pH 7 buffer solution; sweep rate = 500 mV s−1, arrows indicate sweep direction. | ||
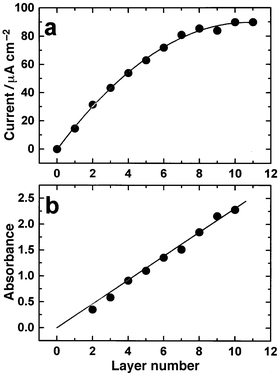 | ||
| Fig. 3 a) Dependence of the cathodic peak current density (jpc) on the number of layers of viologen capped gold particles. Sweep rate = 100 mV s−1. b) Maximum plasmon band absorbance as a function of the number of layers deposited. | ||
In order to gain insight into the structure of the multilayers the growth process was monitored by ex situ UV–VIS spectroscopy and AFM to study the integrity of the nanostructured film. STM imaging of the monolayers and subsequent multilayers proved difficult due to the high resistivity of the alkyl chains. The first monolayer of attached nanoparticles, imaged by AFM, can be clearly seen in Fig. 4a. This layer follows the topography of the substrate, with crystal boundaries still visible as dark areas in the image. After deposition of nine layers of particles and ligand (1) all but the most prominent substrate features have been concealed by the multilayers (Fig. 4b). Cross section analysis of the images shows an increase in the roughness of the sample. These findings indicate that the layers do not build up in a perfect ABAB structure, as simply illustrated in Fig. 1; rather, they form thin organic films containing discrete gold particles separated by the length of the spacer molecules.
 | ||
| Fig. 4 Tapping mode AFM images of: a) a monolayer of gold nanoparticles immobilised on a monolayer of (1) and protected with a monolayer of (1); b) nine layers of gold nanoparticles separated by monolayers of (1) and protected with a monolayer of (1). Image size = 1 μm2, data collected at 2.5 Hz, 256 × 256 resolution. | ||
Whilst AFM imaging can only give information on the surface of the multilayer, UV–VIS spectra of the different films clearly indicate the presence of the gold plasmon band as shown in Fig. 5. For comparison the spectrum of the underivatised gold nanoparticles in toluene solution is shown in the inset. The peak in the spectrum of the toluene solution is due to the plasmon band of the gold nanoparticles (λmax = 526 nm).27 This peak is red-shifted and substantially broadened in the multilayer films giving the films a blue-green appearance in natural light, probably due to coupling of the plasmon excitation on individual particles.28 Comparison of the plasmon peak with the number of layers gives a linear relationship (Fig. 3b) indicating that in each derivatisation step a similar amount of gold is deposited, irrespective of an immersion time longer than the minimum required to form a monolayer.24 If the interior particles were to sinter, as they do in gold hydrosol preparations16 a decrease in the strength of the plasmon band or a shift to a longer wavelength would be observed.17 It can be inferred that the integrity of the particles in the nanostructured material is retained. It is proposed that this is due to the strength of the gold–sulfur bond.
![UV–VIS spectra of 2 to 10 layers of gold nanoparticles linked by (1) and deposited on glass. The inset shows the UV–VIS spectrum of gold nanoparticles in toluene ([Au]total = 1.3 mM, λmax = 526 nm).](/image/article/2000/JM/a902960e/a902960e-f5.gif) | ||
| Fig. 5 UV–VIS spectra of 2 to 10 layers of gold nanoparticles linked by (1) and deposited on glass. The inset shows the UV–VIS spectrum of gold nanoparticles in toluene ([Au]total = 1.3 mM, λmax = 526 nm). | ||
Solid state electron transport properties
Previous conductivity studies have shown that electron transport is dependent on the spacer molecule and does not involve direct metal–metal conduction within the layers (even those containing only four methylene groups); and the resistivity is directly proportional to thickness.17 The linear increase of the plasmon band adsorption with the number of layers (Fig. 3b) shows that a repeat structure is being deposited. However, the plateau in the peak current density observed (Fig. 3a) shows that a limiting current is reached after seven layers of viologen and gold nanoparticles have been deposited. Therefore, only the outer seven layers are accessible for reduction, the inner material appearing inert to redox processes. This can be understood considering that redox processes in the film must involve anion exchange to balance charge.29 In this case, a limiting peak current results from the impermeability of the multilayer to electrolyte, preventing the ionic transport that must occur simultaneously if the electron transfer is to maintain electroneutrality. Internal sintering of nanoparticles to form conducting metal pathways is unlikely since the optical properties of the film correspond to nanoparticles retaining their integrity in the superlattice.17,28 Repeated cyclic voltammetry on films containing eight or more layers showed no significant change in the peak shape, peak potential or peak separation. Therefore, the internal viologen layers, whilst not being electroactive, do not impede electron transfer to the outer layers of the film. This system is therefore analogous to mixed valent viologen crystals containing both dicationic and radical viologens in controlled proportions.30,31 These crystals behave in a similar way to n-doped semiconductors for which a very small addition of radical to the lattice increases its conductivity by several orders of magnitude. These results clearly illustrate conduction by electron hopping from viologen to viologen linker (via the gold nanoparticles in the present case) within the inner layers of material. The kinetics of these processes will be discussed elsewhere.32Electron transport to an external redox couple
The cyclic voltammetry experiments undertaken to analyse the structure of the films focus only on electron transfer through the films rather than to an external aqueous species. The addition of hexaammineruthenium(III) chloride, [Ru(NH3)6]Cl3, to the electrolyte was chosen so as to fulfil the following criteria for the aqueous redox species: 1) it should be cationic, since anions will exchange with the multilayer; 2) it should have a redox potential less positive than that for thiol oxidation; 3) it should have fully characterised, fast (reversible) electron transfer kinetics. Fig. 6 shows three cyclic voltammograms that illustrate the complex electrochemistry of the [Ru(NH3)6]3+/2+ couple and (1). The solid line shows the reduction and subsequent oxidation of the viologen monolayer in the presence of an inert electrolyte. This is compared with the same arrangement but in the presence of the redox active Ru couple (dotted line). The immobilised viologen layer acts as an electron transfer mediator to the aqueous species, but only in one direction, i.e. there is a diode effect. This effect has also been seen previously in surfactant immobilised viologen films,33 in solid-state chemiluminescent systems,34 and in serial mixed valent layers.35 [Ru(NH3)6]3+ reduction can only take place after the viologen has been reduced which can be seen as a shift of the cathodic peak potential of −0.2 V. On the reverse scan the viologen is completely oxidised before the potential at which [Ru(NH3)6]2+ oxidation occurs. Thus the surface is insulating and only permits very slow, irreversible electron transfer across the monolayer, which can only be detected at slow scan rates (<10 mV s−1).36![a) Cyclic voltammogram of the first monolayer of (1) (solid line), 1 mM [Ru(NH3)6]3+ on a clean gold electrode (dashed line), and of 1 mM [Ru(NH3)6]3+ mediated by a monolayer of (1) (dotted line). All experiments were carried out in 0.1 M pH 7 buffer solution at a sweep rate of 50 mV s−1 with no IR correction.](/image/article/2000/JM/a902960e/a902960e-f6.gif) | ||
| Fig. 6 a) Cyclic voltammogram of the first monolayer of (1) (solid line), 1 mM [Ru(NH3)6]3+ on a clean gold electrode (dashed line), and of 1 mM [Ru(NH3)6]3+ mediated by a monolayer of (1) (dotted line). All experiments were carried out in 0.1 M pH 7 buffer solution at a sweep rate of 50 mV s−1 with no IR correction. | ||
A cyclic voltammogram of the electrode covered with the multilayer (10 layers, in Fig. 7) still mediates electron transfer to the [Ru(NH3)6]3+ in solution but drastically inhibits the reoxidation of [Ru(NH3)6]2+. Comparing this with the cyclic voltammogram in Fig. 6 (dotted line) shows the increased capacitance from the nine monolayers of gold nanoparticles, in which the layers exposed to electrolyte act as parallel capacitors. This CV provides further evidence that the internal layers not available for electrochemical reduction do not hinder electron transport since the peak potentials are consistent with those in Fig. 6. It is noteworthy that the onset of electron transfer to the [Ru(NH3)6]3+ complex only occurs at potentials where reduction of the viologen ligand can take place, thus showing that the film acts as an insulating layer until electron hopping can occur through a mixed valent film.
![Cyclic voltammogram of 1 mM [Ru(NH3)6]3+ on a gold electrode mediated by ten layers of (1) and gold nanoparticles. Experiment carried out in 0.1 M pH 7 buffer solution at a sweep rate of 50 mV s−1 with no IR correction.](/image/article/2000/JM/a902960e/a902960e-f7.gif) | ||
| Fig. 7 Cyclic voltammogram of 1 mM [Ru(NH3)6]3+ on a gold electrode mediated by ten layers of (1) and gold nanoparticles. Experiment carried out in 0.1 M pH 7 buffer solution at a sweep rate of 50 mV s−1 with no IR correction. | ||
Conclusions
A novel redox active dithiol has been synthesised which has enabled examination of the electron transfer between gold nanoparticles. The purpose of this work has been to develop assemblies of thiol capped gold nanoparticles as functional materials by examining the relationship between the rate of electron transfer between adjacent metal particles and the availability of low-lying filled and empty molecular orbitals in electroactive organic groups located between particles. Viologen moieties, when incorporated in these multilayers, are addressable electrochemically provided they are within 6–8 layers of the external surface. It has been shown that electron transfer can only take place whilst the viologen is reduced; once oxidised only slow irreversible electron transfer is seen.The results of these investigations show that non-metallic electrical conductivity and electron transport in composite nanostructured materials can be based on electron hopping to well defined energy levels determined by the chemistry of the ligand. In particular this work may cast light on the question of whether assemblies of nanoparticles separated by organic molecules can be regarded as having an electronic band structure. The gating effect observed indicates the additional control provided by a redox-switchable organic moiety located in the spacer molecules which may find novel applications in sub-microelectronic materials.
Acknowledgements
This work was supported by a UK EPSRC studentship (Quota No. 96310533). The support of the University of Liverpool Equipment Fund for the purchase of the AFM/STM microscope is gratefully acknowledged.References
- M. Faraday, Phil. Trans. R. Soc. London, 1857, 147, 145 Search PubMed.
- D. L. Feldheim and C. D. Keating, Chem. Soc. Rev., 1998, 27, 1 RSC.
- G. Schmid, J. Chem. Soc., Dalton Trans., 1998, 1077 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- G. Markovich, D. V. Leff, S. W. Chung, H. M. Soyez, B. Dunn and J. R. Heath, Appl. Phys. Lett., 1997, 70, 3107 CrossRef CAS.
- A. Badia, L. Cuccia, L. Demers, F. Morin and R. B. Lennox, J. Am. Chem. Soc., 1997, 119, 2682 CrossRef CAS.
- R. H. Terrill, T. A. Postlethwaite, C. Chen, C. Poon, A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark, G. Wignall, J. D. Londono, R. Superfine, M. Falvo, C. S. Johnson Jr., E. T. Samulski and R. W. Murray, J. Am. Chem. Soc., 1995, 117, 12537 CrossRef CAS.
- S. W. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098 CrossRef CAS.
- A. P. Alivisatos, Science, 1996, 271, 933 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17 CrossRef CAS.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar, R. L. Whetten, W. G. Cullen, P. N. First, C. Gutierrez-Wing, J. Ascensio and M. J. Jose-Yacaman, J. Phys. Chem. B, 1997, 101, 7885 CrossRef.
- R. Antoine, P. F. Brevet, H. H. Girault, D. Bethell and D. J. Schiffrin, Chem. Commun., 1997, 1901 RSC.
- D. Bethell, M. Brust, D. J. Schiffrin and C. Kiely, J. Electroanal. Chem., 1996, 409, 137 CrossRef CAS.
- R. P. Andres, J. D. Bielefeld, J. I. Henderson, D. B. Janes, V. R. Kolagunta, C. P. Kubiak, W. J. Mahoney and R. G. Osifchin, Science, 1996, 273, 1690 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS.
- M. D. Musick, C. D. Keating, M. H. Keefe and M. J. Natan, Chem. Mater., 1997, 9, 1499 CrossRef CAS.
- M. Brust, D. Bethell, C. J. Kiely and D. J. Schiffrin, Langmuir, 1998, 14, 5425 CrossRef CAS.
- C. N. R. Rao, J. Mater. Chem., 1999, 9, 1 RSC.
- M. D. Musick, D. J. Pena, S. L. Botsko, T. M. McEvoy, J. N. Richardson and M. J. Natan, Langmuir, 1999, 15, 844 CrossRef CAS.
- M. Antonietti and C. Goltne, Angew. Chem., Int. Ed. Engl., 1997, 36, 910 CrossRef.
- P. M. S. Monk, The Viologens, Wiley, Chichester, 1998. Search PubMed.
- M. Brust, R. Etchenique, E. J. Calvo and G. J. Gordillo, Chem. Commun., 1996, 1949 RSC.
- C. A. Goss, D. H. Charych and M. Majda, Anal. Chem., 1991, 63, 85 CrossRef CAS.
- L. Zhang, T. Lu, G. W. Gokel and A. E. Kaifer, Langmuir, 1993, 9, 786 CrossRef CAS.
- J. F. Rodriguez, T. Mebrahtu and M. P. Soriaga, J. Electroanal. Chem., 1987, 233, 283 CrossRef CAS.
- C. A. Widrig and M. Majda, Langmuir, 1989, 5, 689 CrossRef CAS.
- P. Mulvaney, Langmuir, 1996, 12, 788 CrossRef CAS.
- T. Baum, D. Bethell, M. Brust and D. J. Schiffrin, Langmuir, 1999, 15, 866 CrossRef CAS.
- H. C. DeLong and D. A. Buttry, Langmuir, 1990, 6, 1319 CrossRef CAS.
- B. Emmert, G. Jungck and H. Haffner, Chem. Ber., 1924, B57, 1792 Search PubMed.
- D. R. Rosseinsky and P. M. S. Monk, J. Chem. Soc., Faraday Trans., 1994, 90, 1127 RSC.
- D. I. Gittinset al., paper in preparation..
- M. Gomez, J. Li and A. E. Kaifer, Langmuir, 1991, 7, 1797 CrossRef CAS.
- K. M. Maness, R. H. Terrill, T. J. Meyer, R. W. Murray and R. M. Wightman, J. Am. Chem. Soc., 1996, 118, 10609 CrossRef CAS.
- H. Masui and R. W. Murray, J. Electrochem. Soc., 1998, 145, 3788 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 1980. Search PubMed.
Footnote |
| † Basis of a presentation given at Materials Chemistry Discussion No. 2, 13–15 September 1999, University of Nottingham, UK. |
| This journal is © The Royal Society of Chemistry 2000 |