Speciation of arsenic in edible algae by bi-dimensional size-exclusion anion exchange HPLC with dual ICP-MS and electrospray MS/MS detection
Shona McSheehy and Joanna Szpunar
CNRS EP132, Hélioparc, 2, rue Pr. Angot, 64053, Pau-Pyrénées, France. E-mail: joanna.szpunar@univ-pau.fr
First published on UnassignedUnassigned7th January 2000
Abstract
Anion-exchange (AE) high-performance liquid chromatography (HPLC) with ICP-MS detection was revisited for speciation of arsenic in biomaterials with particular attention given to arsenic-containing ribosides (arsenosugars) in seaweeds. Signal identification by retention time matching and spiking experiments was found to be difficult because of the co-elution of the ribosides with other arsenic species [As(III), monomethylarsonic acid], the medium- and long-term irreproducibility of retention times of arsenic species and an uncontrolled matrix effect on the retention times. Size-exclusion (SE) HPLC was proposed for the fractionation of organoarsenic species and matrix removal prior to detection of arsenosugars by AE-HPLC followed by signal identification by spiking experiments and retention time matching. The results were compared with those obtained by pneumatically-assisted electrospray tandem MS (ESI MS/MS) of the SE-HPLC fractions. A number of 10 commercially available edible algal food samples of different origins available on the French market were investigated. The approaches developed for the identification and determination of arsenosugars (SE-AE-HPLC-ICP-MS and SE-HPLC-ESI MS/MS) were discussed using the example of Hizikia fusiforme.
Introduction
Arsenic has the notoriety of being a toxic element but it is also widely established that its toxicity is critically dependent on the species.1 Marine plants and animals are known to contain unusually high levels of arsenic (1–10 mg g−1 wet weight) despite the few thousand times lower concentration of this element (low µg L−1) present in sea water.2,3 This particular capacity to absorb oceanic arsenate by organisms that often become food products raises concerns in countries where algae and other seafood are the major constituent of the human diet.Arsenic biotransformations in marine life are known to lead to a wide range of organoarsenic compounds. Arsenobetaine (trimethylarsonioacetate) is the most abundant arsenic species in marine animals whereas macroalgae and some bivalves contain a class of dimethylarsinoyl ribosides known under the trivial name of arsenosugars.1–3 Until recently, the development of analytical methodology for the determination of arsenosugars in edible resources might have seemed to be academic since this class of compounds, like arsenobetaine, have generally been believed to be innocuous to man.4 However, humans are apparently able to convert arsenosugars to dimethylarsinic acid (DMAA)5 and some recent studies indicate that DMAA has the potential to be a human carcinogen.6 Also recently, a case of intoxication by organic arsenic was reported following the consumption of a bird's nest, as it is a delicacy in South Asian countries.7
To date, the most significant work on the characterization of arsenosugars in marine life has been carried out by Edmonds and co-workers who described a number of arsenic-containing ribosides from different brown algae (Sargassum,Ecklonia,Hizikia)8–10 and kidney of the giant clam Tridacna.11 They isolated a number of organoarsenic compounds by consecutive preparative gel, anion-exchange (AE) and thin layer chromatography prior to identification of arsenosugars by 1H and 13C NMR spectroscopy. The obvious disadvantage of the last is the need of the availability of milligram quantities of well purified compounds for analyses that require the processing of several kilograms of samples and use of tens of liters of solvents for extraction. Moreover, the application to terrestrial organisms that contain 10-times lower concentrations of organoarsenic species seems to be even more difficult. There is a need for a rapid and sensitive analytical approach to the characterisation of arsenoribosides. For this purpose the coupling HPLC-ICP-MS was investigated.12–15
The HPLC separation mechanisms have the advantage that arsenosugars are readily and differently ionized at different pHs owing to the presence of a number of functional groups such as Me2As(O)- and -SO3H. The ions formed (cations due to the protonation of the dimethylarsinoyl moiety at low pH and anions due to the dissociation of the -SO3H groups at neutral pH) consist of hydrophobic ion-pairs with a suitable counter-ion that are then chromatographed by reversed-phase (RP) HPLC or size-exclusion (SE) HPLC.12–15 Standards are required for signal identification by retention time matching. A number of problems rise due to the retention time irreproducibility, common in the presence of a sample matrix of which some constituents may compete for arsenosugar ions with molecules of the ion-pairing reagent.16 Also, not only is the availability of arsenosugar standards problematic but the efficiency of a chromatographic separation may turn out to be insufficient to separate all the organoarsenic compounds from each other. Consequently, a risk occurs that the spike of one compound will match the retention time of another one, leading to misidentification. This risk can be reduced by the use of bi-dimensional chromatography. An alternative is the use of molecular MS that requires 100–1000 times less sample for analysis than NMR. Also, the degree of purification required becomes less crucial.
Fast atom bombardment (FAB) tandem MS in the negative and positive modes has been proposed for the characterisation of arsenosugars and applied to a partially purified Sargassum lacerifolium algal extract.17 A much better sensitivity can be achieved by pneumatically assisted electrospray tandem MS (ESI-MS/MS) which also offers the possibility of on-line chromatographic detection;18,19 an example of a cation-exchange chromatogram showing an arsenosugar peak in the NBS SRM 1566a Oyster tissue has been reported.19 Electrospray ionization suffers from the problem of matrix suppression effect on the signal and the compatibility of the chromatographic mobile phase with the ionization conditions. In particular, the use of ion-pairing reagents seems to be disastrous requiring an alternative to ion-pairing chromatography separation approaches.20
The objectives of this work are: (i) to develop an analytical methodology for the routine screening and unambiguous identification of arsenosugars in edible algae by HPLC-ICP-MS, and (ii) to investigate the use of ESI-MS/MS for the identification of arsenosugars in algae.
Experimental
Apparatus
Chromatographic separations were carried out using a Hewlett Packard series 1100 pump (Hewlett-Packard, Waldbronn, Germany) as the sample delivery system. Injections were carried out using a Model 7725 injection valve with a 100 µL injection loop (Rheodyne, CA, USA). All the connections were made of PEEK tubing (id 0.17 mm).The ICP-MS instrument used in this work was the Elan 6000 (PE-SCIEX, Thornhill, ON, Canada). The sample introduction system used included a Ryton™ spray chamber fitted with a cross flow nebulizer. For non-chromatographic preliminary optimisation studies, the samples were fed by means of a Minipuls 3 peristaltic pump (Gilson, France) that also served for draining the spray chamber. Chromatographic signals were processed using the Turbochrom4™ software (Perkin-Elmer). All signal quantifications were made in the peak area mode.
The electrospray MS instrument used was a Perkin-Elmer SCIEX (Thornhill, ON, Canada) API 300 pneumatically-assisted electrospray (ion-spray) triple-quadrupole mass spectrometer. Samples were introduced using a syringe pump (Harvard Apparatus, South Natick, MA, USA). Ultrasonic extraction was performed using a Branson 1210 ultrasonic cleaner (Danbury, CT, USA). The supernatant was separated by centrifugation using a Model Hettich Universal 16 (Tuttlingen, Germany). The rotary evaporator used for the elimination of methanol was a Heidolph VV Mikro (Germany). Lyophilization was carried out using a Model LP3 lyophilizer (Jouan, France).
Materials and reagents
Size-exclusion chromatographic separations were carried out on a Superdex Peptide HR 10/30 column (30 cm × 10 mm × 13 µm) (Pharmacia, Uppsala, Sweden). Anion-exchange separations were carried out using a Supelcosil SAX1 (250 × 4.6 mm × 5 µm) (Supelco, Bellefonte, PA, USA) column with a Supelguard SAX1 guard column.Analytical reagent-grade chemicals were used throughout unless otherwise stated. Methanol (Sigma-Aldrich, St. Quentin Fallavier, France) was of LC grade. Water was purified to 18.2 MW resistance using a Milli-Q water purification system (Millipore, Bedford, MA, USA). The 25 mM phosphate buffer for anion-exchange chromatography was prepared by mixing 60 mL of 0.2 M NH4H2PO4 with 5 mL of 0.2 M (NH4)2HPO4 and making up the solution to 500 mL. The 5 mM phosphate buffer was prepared by diluting the 25 mM buffer with water. For size exclusion chromatography, a 1% (v/v) solution of acetic acid was prepared. The buffers were degassed by sonication for 20 min before starting chromatography.
Standards and samples
Standard stock solutions at a concentration of 1.00 mg mL−1 were prepared by dissolving the respective compound in water. Arsenic(III) and arsenic(V) standards were prepared from sodium arsenate and sodium arsenite (Sigma Aldrich, St. Quentin Fallavier, France) in water. The four dimethylarsinoyl-riboside derivatives: 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropanesulfonic acid (referred to later as sugar A), 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropylene glycol (sugar B), 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropyl hydrogen sulfate (sugar C) and 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropyl 2,3-hydroxypropyl phosphate (sugar D) were kindly donated by Professor J. Edmonds (De Montfort University, Leicester, UK).Arsenobetaine was a gift from Professor W. Cullen (UBC, Vancouver, Canada). Standard solutions of the other arsenic compounds were a gift from Dr. Erik Larsen (Danish Veterinary and Food Administration, Søborg, Denmark). A secondary stock solution of 1 mg mL−1 of each of the compounds was prepared for HPLC-ICP-MS analysis. Working solutions were prepared on the day of analysis by the appropriate dilution of the stock solutions with water. The stock solutions were kept in the fridge at 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C in the dark.
°C in the dark.
A number of dried algal food-products commercially available on the French market was supplied by the regional D.G.C.C.R.F. agency (Direction Générale de la Concurrence et de la Consommation de la Republique Française) of the French Ministry of Finances and Economy (Bordeaux, France). The samples originated from France, Spain, Iceland, Japan and China and were available as food products imported from the Far East or as food products ground into powders. Commercial products were kept in their packages away from direct sunlight in the laboratory. Powdered samples were transferred to plastic bottles and stored in the same fashion. The commercial products were soaked in cold water for ten minutes before preparation for consumption in soups or salads. The list of samples investigated in this work is given in Table 2.
Procedures
°C in a water bath. Total arsenic was determined by GFAAS. Mineral (inorganic) arsenic was determined on a separate portion of algae according to the procedure of Whyte and Englar,21 which is based on the separation of inorganic arsenic as AsCl3 by distillation from organoarsenic compounds.°C by means of a rotary evaporator. The residue after evaporation to dryness was dissolved in 10 mL of water prior to analysis.For SE-AE-HPLC the fractions were collected from 6 injections of 100 µL of extract, the eluate was frozen and lyophilized, and the residue after lyophilization was dissolved in 600 µL water. The relevant arsenic fractions were collected following SE-HPLC for subsequent AE-HPLC under anion-exchange conditions.
| HPLC conditions— | |
| Anion exchange column | Supelcosil SAX1 |
| Mobile phase | 5 to 30 mM phosphate buffer (pH 6) within 22 min |
| Flow rate | 1.0 mL min−1 |
| Size-exclusion column | Superdex Peptide HR 10/30 |
| Mobile phase | 1% aq. acetic acid, pH 3 |
| Flow rate | 0.6 mL min−1 |
| ICP MS conditions— | |
| Forward power | 1100 W |
| Nebulizer gas flow rate | 1.05 L min−1 |
| Isotope monitored | 75As |
| Dwell time | 60 ms |
| ESI MS/MS conditions— | |
| Orifice | 20 V |
| Ionspray voltage | 4100 V |
| Scan range | 70–500 u within 8.6 s |
| Dwell time | 1 ms |
| Step size | 0.05 u |
| MS CID MS conditions— | |
| Parent ion | 483, 409 |
| Collision energy | 20 eV |
| Product ion scan range | 70–500 within 7.17 s |
| Dwell time | 5 ms |
| Step size | 0.3 u |
| Multiplier voltage | 2400 V |
Electrospray MS/MS conditions
The optimization of the ESI-MS/MS conditions was carried out using 10 µg ml−1 of each of the arsenosugar solutions in a water–methanol mixture (70∶30 v/v) containing 0.6% of 0.1 M hydrochloric acid. The optimum operating conditions are given in Table 1. An analysed sample solution was prepared from a size-exclusion chromatographic fraction, lyophilized and redissolved in water as described above, by adding an appropriate amount of methanol and hydrochloric acid.Results and discussion
Determination of the total arsenic and the mineral arsenic
The algae samples analysed in this study had been withdrawn from the French market on the basis of the too high (>3 mg kg−1) concentrations of the mineral arsenic. The mineral arsenic was determined according to the procedure of White and Englar21 that is based on the separation of inorganic arsenic as AsCl3 from organoarsenic compounds by distillation. The total arsenic concentration was determined by GFAAS after mineralization of the sample as described in the procedure. The accuracy of the method was verified by the analysis a reference algae (Laminaria digitata). The mineral arsenic was found to be 72 ± 7 mg kg−1 (8 analyses) compared with the recommended value of 61 mg kg−1 whereas the total arsenic was 124 ± 6 mg kg−1 (5 analyses) compared with the recommended value of 145 mg kg−1.The values for the total arsenic and for the mineral arsenic found in the samples investigated are summarized in Table 2. The total concentrations of arsenic found in the samples varied from 3.5 to 134 mg g−1 out of which the “mineral" arsenic constituted between 36–74%. The rest is supposed to be arsenosugars on the basis of earlier literature reports.8–11 In particular two samples, Hizikia and Laminaria, were found to contain ca. 10-times higher concentration of organic arsenic in comparison with the other algae products and were used as model samples for the development of the analytical procedures.
| Sample | Species of algae | Origin of algae | Total As/mg kg−1 | Mineral As/mg kg−1 |
|---|---|---|---|---|
| aNot edible sample. | ||||
| A | Fucales and Laminares | Spain | 8.4 | 3.3 |
| B | Hizikia fusiforme | Japan | 54 | 34 |
| C | Hizikia fusiforme | Japan | 45 | 22 |
| D | Himanthalia | Brittany | 3.6 | 2.4 |
| E | Laminaria | Brittany | 134 | 62 |
| F | Laminaria | Iceland | 30 | 18 |
| G | Palmaria palmata | Brittany | 5.3 | 1.9 |
| H | Porphyra umbilicalis | Brittany | 5.4 | 3 |
| Ia | Sargassum lacerifolium | Australia | Not analysed | Not analysed |
| J | Not specified | China, Hong Kong | 3.5 | 2.6 |
| K | Undaria Pinnatifada | Japan | 5.6 | 4 |
Species-specific analysis for organoarsenic compounds in algae
The prerequisite of applying a chromatographic method to the detailed characterization of the organic arsenic is its extraction into an aqueous phase. Methanol has commonly been used for the extraction of arsenic from fresh seaweeds of which the water content exceeds 90%.8–11 Since the products we needed to analyse were dry, the use of a mixture of water–methanol (50∶50 v/v) was found optimum to give the maximum extraction yield of arsenic that exceeded 85%.The fact that organoarsenic compounds are readily and differently ionized at different pHs makes ion-exchange a natural choice as the HPLC separation mechanism. Anion-exchange-HPLC-ICP-MS is considered as an established technique for speciation of arsenic; isocratic elution in neutral media was recommended by Larsen in an excellent working method paper for the separation of the five major arsenic species [As(III), As(V), MMAA, DMAA, and arsenobetaine].15 For the analysis of samples containing significant amounts of arsenosugars the use of anion-exchange and cation-exchange in parallel was recommended to avoid the identification problems due to the possible overlaps among organoarsenic compounds occurring in each of the techniques.23–26
The chromatographic purity of signals can be improved by using chromatographic techniques with different separation mechanisms not in parallel but in series. The limitations of AE-HPLC for speciation of arsenic in arsenosugar-rich algae were investigated first; the possibility of improving the purity of organoarsenic compounds in this technique was examined by preceding the anion-exchange separation by size-exclusion chromatography. The optimization and performance of the methodology is discussed below for the example of a Hizikia fusiforme algae. It was reported elsewhere to contain ca. 50% of the total arsenic as arsenate, the remainder being arsenosugars,10 which is roughly in agreement with our observation.
Speciation of arsenosugars by anion-exchange HPLC-ICP-MS
The application of the isocratic elution conditions at neu-tral24–26 or alkaline23 pH, which are usually recommended for speciation of arsenic in biological materials to the separation of arsenosugars, leads to the elution of the latter in two fractions: arsenosugar B which does not dissociate under these conditions and elutes with the void, and the other arsenosugars, A, C, D, which interact with the column and elute together giving one signal in HPLC-ICP-MS (chromatogram not shown). The first peak overlaps with As(III) and the second one with MMAA, which makes an unambiguous detection of arsenosugars in real-world samples impossible. Elution using the concentration gradient of the buffer in neutral media (pH 6.0) was therefore optimized to achieve the baseline resolution of arsenosugars and to discriminate them from other As species potentially present in a sample. Various linear gradients ending in a buffer concentration of 15, 20, 25 and 30 mM at times varying from 15 to 30 min were investigated with the objective of achieving the baseline separation of arsenosugar compounds in the minimum time. This was achieved using a linear gradient from 5 to 25 mM of the buffer within 22 min. As the column efficiency decreased with the long term use of the column, the concentration of the final buffer was decreased to 20 mM and then to 15 mM to maintain the baseline resolution of arsenosugar standards.A typical chromatogram obtained under the optimized conditions is shown in Fig. 1a. It shows the baseline separation of the four arsenosugars investigated. The arsenosugars A and D are free from overlaps with other organoarsenic compounds but arsenosugar C overlaps with MMAA. Arsenosugar B elutes in the void prior to As(III) and arsenobetaine, but no baseline resolution among these signals could be achieved.
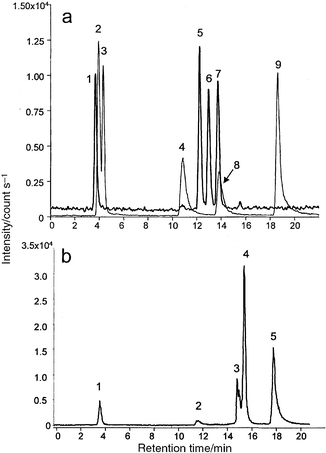 | ||
| Fig. 1 Anion-exchange HPLC chromatograms under the optimized conditions. a, Arsenic standards: bold line, arsenosugars; regular line, other As species. Peak identification: 1, arsenosugar B (60 ng mL−1); 2, As(III) (100 ng mL−1); 3, arsenobetaine (100 ng mL−1); 4, DMAA (100 ng mL−1); 5, arsenosugar D (130 ng mL−1); 6, arsenosugar A (67 ng mL−1); 7, arsenosugar C (130 ng mL−1); 8, MMAA (100 ng mL−1); 9, As(V) (100 ng mL−1). b, Extract of Hizikia fusiforme (diluted by 100 times). Peak identification: 1, arsenosugar B or As(III); 2, DMAA; 3, arsenosugars D + A or MMAA; 4, arsenosugar C; 5, As(V). | ||
A typical chromatogram of an algae extract is shown in Fig. 1b. It shows 4–5 signals of which the identification by retention time matching is problematic. Despite the 20 to 100-fold dilution of the lyophilized extract prior to injection, the retention times of signals in the sample do not correspond to those in the standard solutions. A reason may be that other arsenic species are present in the sample extract. However, a more likely explanation is that the algae matrix contains constituents that modify the stationary phase or interact with the analyte species thus changing their retention behaviour. Indeed, the retention time was actually found to be stable (within 1–2%) for a given column on the same day but column-to-column or day-to-day variations can be significant, requiring spiking experiments for compound identification.
The spiking experiments can confirm the identity of peaks 2-DMAA, 4-arsenosugar C and 5-As(V). However, the signals from MMAA, arsenosugar A and arsenosugar D overlap with a signal observed in the chromatogram of the sample (peak 3) rendering the identification of the last impossible. Note that, in comparison with the standards, MMAA overlaps with arsenosugars A and D and not with compound C; an observation which emphasizes again the extreme care with which the retention time data should be interpreted. As(III) is undistinguishable from arsenosugar B even if a slight difference between the retention times was noted in the chromatogram of standards (Fig. 1).
In summary, anion-exchange chromatography of algal extracts suffers from insufficient resolution, which results in overlaps of signals from different arsenic species, making their identification impossible. Moreover, the retention time and the resolution are dependent on the long term use and the actual state of the column. These facts make HPLC-ICP-MS inappropriate for speciation of arsenic in algal extracts. In order to avoid the matrix effect on the shift in retention times and on the overlap of the cluster of arsenosugars by MMAA, it was judged necessary to introduce an additional chromatographic separation step prior to anion-exchange chromatography which would result in bi-dimensional chromatography.
Speciation of arsenosugars by SE-HPLC-ICP-MS
Gel chromatography using Sephadex LH-20 stationary phase was the first step in the classical procedures for the isolation of arsenosugars from marine algae.8–11 Analytical SE-HPLC-ICP-MS with mobile phases fairly rich (25 mM) in an ion-pairing reagent was proposed for speciation of organoarsenic, including arsenosugars in seeweeds,27 dogfish,12 urine,13 and marine fauna.14 Since, in our case, the presence of ion-pairing reagents was considered to be undesirable in view of the subsequent AE-HPLC or ES-MS/MS, it was attempted to investigate SE-HPLC of organoarsenic compounds with pure or slightly acidified water, as demonstrated elsewhere for organoselenium compounds.28 The choice of the column was made on the basis of the separation range and size of organoarsenic species. A column with the minimum available separation range (exclusion limit 10 kDa) initially designed for the separation of small peptides was selected.Fig. 2a and b shows a SE-HPLC-ICP-MS chromatogram of arsenic standards under the optimum separation conditions. It can be seen that organoarsenic compounds elute within 4 fairly distinct separated envelopes. The first one contains arsenosugars A, C, D (peaks 1–3), the second one contains As(V), MMAA, DMAA (peaks 4–6) and arsenobetaine (not shown), the third contains tetramethylarsonium (TMA), arsenocholine, arsenosugar B (peaks 7–9) and trimethylarsine oxide (not shown), and the last one As(III) (peak 10). The most important feature from the point of view of the subsequent work is that a fraction containing arsenosugars A, C, D can be readily separated from the rest of organoarsenic species and probably from many matrix constituents, which makes SE-HPLC the ideal technique to precede AE-HPLC for speciation analysis of organoarsenic in algal food products.
 | ||
| Fig. 2 Separation of organoarsenic compounds by size-exclusion chromatography with ICP-MS detection: a, arsenosugar standards; b, other species; c, algal extract (Hizikia fusiforme). Standards were injected individually. Peak identification: 1, arsenosugar D; 2, arsenosugar A; 3, arsenosugar C; 4, As(V); 5, DMAA; 6, MMAA; 7, arsenosugar B; 8, TMA cation; 9, arsenocholine; 10, As(III). Arsenobetaine (not shown) elutes between peaks 5 and 6. TMA oxide (not shown) overlaps peaks 8 and 9. The vertical dashed line discriminates the arsenosugar fraction. The dotted lines (panel c) discriminate the fractions collected for the identification of peaks discussed further in the text. | ||
Fig. 3 shows SE-HPLC-ICP-MS chromatograms of extracts of algae samples listed in Table 2. None of the products contains As(III), which is considered the most toxic form of the element. In the majority of cases (7 out of 10) four signals can be distinguished. They are grouped in two pairs of which the first corresponds to the arsenosugars (A, C, D) fraction whereas the second one contains other arsenic compounds. In two cases the sugar envelope is maintained but the other arsenic species elute as one peak. In three cases only two signals can be seen; one corresponding to the arsenosugar fraction and the other to other arsenic species. The precision of the elution volume of the arsenosugar envelope is rather good except for the three samples where only two peaks can be seen. It is worth noting that the ratio of As(V) to the total As, estimated from the SE-HPLC-ICP-MS chromatograms, is two times lower than that resulting from the direct total and inorganic As measurement. The reason for this remains for the time being unknown.
 | ||
| Fig. 3 Speciation of arsenic in algae extracts by SE-HPLC-ICP-MS. The letters A–K correspond to samples listed in Table 2. No unambiguous peak identification is possible at this stage. | ||
SE-HPLC allows the separation of arsenosugars from the other arsenic species examined, but the risk of overlaps with other arsenic compounds not present among the standards investigated is judged to be too important for SE-HPLC-ICP-MS to be used alone to quantify the level of arsenosugars present in a sample. It is necessary to verify the chromatographic purity of signals observed in SE-HPLC-ICP-MS, which can be achieved by extracting the relevant fraction and subjecting it to a chromatographic technique with a complementary separation mechanism, viz., anion-exchange HPLC. The development of such an approach is presented below using the example of the algae Hizikia fusiforme.
Speciation of arsenic compounds in algal extracts by bidimensional SE-AE-HPLC-ICP-MS
The fractions collected for AE-HPLC-ICP-MS were marked in the SE-HPLC-ICP-MS chromatogram (cf.Fig. 2c). The collected eluate was frozen, lyophilized and redissolved in water as described under Procedures. The chromatograms of the four fractions including those obtained for the purpose of signal identification by spiking experiments are shown in Fig. 4. | ||
| Fig. 4 Identification of arsenic species in algae extracts by bi-dimensional SE-AE-HPLC-ICP-MS and spiking experiments. The SE-HPLC-ICP-MS chromatogram and the fraction description is given in Fig. 4c. a, AE-HPLC-ICP-MS chromatograms of fraction I; b, chromatograms of fraction II; c, chromatograms of fraction III; d, chromatogram of fraction IV. Bold line, original chromatogram of a SE-HPLC fraction. Thin line, chromatogram of the same fraction spiked with an appropriate standard. Peak identification: 1,2, As(III), arsenosugar B or unknown; 3, DMAA; 4, unidentified; 5, arsenosugar D; 6, MMAA or arsenosugar A; 7, arsenosugar C or unknown; 8, unknown; 9, As(V). See also the discussion in the text. | ||
Fig. 4a and b (arsenosugar fraction) demonstrates that the fractionation by SE-HPLC prior to AE-HPLC improves considerably the chromatographic resolution of the latter. Indeed, the retention times of the sugars D, A, and C are now separated by the same intervals as in the case of the chromatogram of standards (Fig. 1a) in contrast to that obtained for the crude algal extract (Fig. 1b). Unlike when standards are used, the baseline resolution is not achieved, probably because of the presence of smaller quantities of other non-identified arsenic species.
For sugar C, the matching of the retention time with the spike is unconvincing. A detailed analysis of the chromatogram indicates that, in the sample chromatogram, this peak has a shoulder on its ascending part and it is the retention time of this shoulder that matches that of sugar C. The chromatogram of the spike shows a shoulder on the descending part of the peak of arsenosugar C which corresponds to the unknown peak in the sample. It is concluded that peak 7 may indeed contain some of sugar C, but it is another unidentified As species that is responsible for producing peak 7.
In summary, fraction I contains sugar D with an admixture of sugar A, the latter at a concentration 5–10 times lower. The peak 1 in the void and peak 2 close to it remain unidentified. Another unknown signal precedes the cluster of arsenosugars D, A, C.
Fig. 4b shows the close-to-baseline separation of peaks 4–8; the spikes with arsenosugars D, A and C allow the positive identification of some of these compounds. Since MMAA cannot be present in the second fraction (panel top left) it is again tempting to attribute the peak at this retention time in the chromatogram to sugar A, which is confirmed by retention time matching with the appropriate standard (panel top right). A trace of sugar D is still present (note that the resolution of arsenosugars D, A and C is now as good as that in the standard solution). In fraction II the signal from arsenosugar A is more intense than that of D. The bottom right panel shows unambiguously the presence of arsenosugar C. It elutes slightly after sugar A from the SE column and is perfectly matched by the spike of the standard.
There are two more unidentified peaks; one (peak 4) preceding and one (peak 8) following the arsenosugar envelope, which are not present in the spiked standards.
Signal identification in SE-HPLC-ICP-MS by electrospray MS/MS
An alternative to bi-dimensional chromatography for signal identification which is limited by the availability of standards is the use of a species-selective detection technique such as electrospray MS. The direct ESI-MS analysis of a crude extract is usually not successful because of a matrix which is too complex. The latter can be simplified by SE-HPLC taking advantage of the early elution of arsenosugars D, A and C. | ||
| Fig. 5 Electrospray MS of SE-HPLC fractions of algae extracts obtained as indicated in Fig. 2c. a, Fraction I; b, fraction III. No As-containing peaks could be identified in fractions III and IV. | ||
Molecular mass spectra are much simpler in comparison with the FAB spectra reported elsewhere.17 Nevertheless, arsenic having only a single stable isotope means that the attribution of a peak to an arsenic containing species requires an á priori hypothesis on the identity of a compound expected to be found. The sensitivity of ESI-MS is much higher than that of FAB-MS. Approximately 1–5 ng of an arsenosugar are sufficient for the acquisition of spectra shown in Fig. 5 in comparison with 600–1700 ng necessary for an equivalent quality FAB-MS spectrum.17 Note that the sensitivity in both MS techniques is negatively affected by the matrix.
The ESI-MS spectrum of fraction I shows a signal corresponding to arsenosugar D (Fig. 5a) whereas in fraction II the signal corresponds to arsenosugar C (Fig. 5b). No traces of sugar A were detected but, since the intensity of signals for sugars D and C is already very poor, the signal from sugar A would be lost in the noise. Since these signals show a fairly low signal to-noise intensity their identity should be verified by the product ion scan of the collision induced dissociation (CID) of the molecular ion.
No signals with an intensity above three times the noise level could be detected in fraction III and IV. Although the intensity of the MMAA and DMAA responses in ESI-MS is higher than that of arsenosugars19 these fractions are likely to contain a rich matrix in comparison with fractions I and II and therefore signal depression follows.
 | ||
| Fig. 6 Collision-induced dissociation (product ion scan) mass spectra of signals suspected to originate from arsenosugars in Fig. 5 (marked with an arrow). Panels a and b correspond to the respective panels in Fig. 5. | ||
ESI-MS/MS allows the unambiguous identification of the presence of arsenosugar D in fraction I and arsenosugar C in fraction II. It is faster than SE-AE-HPLC-ICP-MS and does not require standards. However, the detection limits are a factor of 10 worse than in HPLC-ICP-MS,30 the sensitivity of ESI-MS/MS being affected by the matrix composition.
Conclusions
The identification of arsenic signals in HPLC-ICP-MS chromatograms of algal extracts by retention time matching with standards is unreliable since this parameter is strongly affected by the current state of the column and the composition of the matrix. Spiking experiments can correct for shifts in the retention time of species of interest but the unambiguous positive signal identification on this basis remains illusory. The large number of organoarsenic species present in marine life renders highly probable the risk of the occurrence of two or more As compounds having the same retention time, especially when the chromatographic resolution diminishes in the presence of matrix components. Bi-dimensional chromatography using two different separation mechanisms offers a means of verification of the chromatographic purity of eluted compounds, improves the resolution and reduces the risk of overlaps allowing a more reliable signal identification by spiking experiments. Electrospray MS is an attractive alternative but the potential of this technique in the case of arsenic is hampered by its being mono-isotopic and by the resulting difficulty of the unambiguous attribution of a peak in the mass spectrum to an arsenic species, even in the case of extracts fractionated by SE-HPLC. Tandem MS is therefore mandatory for the ultimate confirmation of the identity of arsenosugars present.Acknowledgements
S. McSheehy acknowledges a Marie Curie fellowship (Grant No SMT-4CT-982232). The authors thank J. S. Edmonds (de Montfort University, Leicester) and R. Lobinski (CNRS, Pau) for valuable discussions. M. Marcinek (Warsaw University of Technology, Warsaw) is acknowledged for help with some measurements.References
- J. S. Edmonds and M. Morita, Pure Appl. Chem., 1992, 64, 575 CrossRef.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713 CrossRef.
- K. A. Francesconi and J. S. Edmonds, Oceanogr. Mar. Biol. Annu. Rev., 1993, 31, 111 Search PubMed.
- V. W. Lai, W. R. Cullen, C. F. Harrington and K. J. Reimer, Appl. Organomet. Chem., 1997, 11, 797 CrossRef CAS.
- X. C. Le, W. R. Cullen and K. J. Reimer, Clin. Chem., 1994, 40, 617 Search PubMed.
- S. Yamamoto, Y. Konishi, T. Murai, M. A. Shibata, T. Matsuda, K. Kuroda, G. Endo and S. Fukushima, Appl. Organomet. Chem., 1994, 8, 197 CrossRef CAS.
- K. V. Luong and L. T. Nguyen, Am. J. Med. Sci., 1999, 317, 269 Search PubMed.
- K. A. Francesconi, J. S. Edmonds, R. V. Stick, B. W. Skelton and A. H. White, J. Chem. Soc., Perkin Trans. 1, 1991, 2707 RSC.
- J. S. Edmonds and K. A. Francesconi, J. Chem. Soc., Perkin Trans. 1, 1983, 2375 RSC.
- J. S. Edmonds, M. Morita and Y. Shibata, J. Chem. Soc., Perkin Trans. 1, 1987, 577 RSC.
- K. A. Francesconi, J. S. Edmonds and R. V. Stick, J. Chem. Soc., Perkin Trans. 1, 1992, 1349 RSC.
- M. Morita and T. Shibata, Anal. Sci., 1987, 3, 575 Search PubMed.
- M. Morita and T. Shibata, Anal. Sci., 1989, 5, 107 Search PubMed.
- M. Morita and T. Shibata, Anal. Chem., 1989, 61, 2116 CrossRef CAS.
- E. H. Larsen, Spectrochim. Acta, Part B, 1998, 53, 253 CrossRef.
- W. Goessler, W. Maher, K. J. Irgolic, D. Kuehnelt, C. Schlagenhaufen and T. Kaise, Fresenius' J. Anal. Chem., 1997, 359, 434 CrossRef CAS.
- S. A. Pergantis, K. A. Francesconi, W. Goessler and J. E. Thomas-Oates, Anal. Chem., 1997, 69, 4931 CrossRef CAS.
- J. J. Corr, J. Anal. At. Spectrom., 1997, 12, 537 RSC.
- J. J. Corr and E. H. Larsen, J. Anal. At. Spectrom., 1996, 11, 1215 RSC.
- H. ChassaigneR. Lobinski, personal communication..
- J. N. C. Whyte and J. R. Englar, Botanica Marina, 1983, 26, 159 Search PubMed.
- Y. Shibata and M. Morita, Appl. Organomet. Chem., 1992, 6, 343 CrossRef CAS.
- E. H. Larsen, Fresenius' J. Anal. Chem., 1995, 352, 582 CrossRef CAS.
- W. Goessler, W. Maher, K. J. Irgolic, D. Kuehnelt, C. Schlagenhaufen and T. Kaise, Fresenius' J. Anal. Chem., 1997, 359, 434 CrossRef CAS.
- W. Goessler, A. Rudorfer, E. A. Mackey, P. R. Becker and K. J. Irgolic, Appl. Organometal. Chem., 1998, 12, 491 CrossRef CAS.
- Z. Slejkovec, J. T. van Elteren and A. R. Byrne, Talanta, 1999, 49, 619 CrossRef CAS.
- J. Yoshinaga, Y. Shibata, T. Horiguchi and M. Morita, Accred. Qual. Assur., 1997, 2, 154 CrossRef CAS.
- C. Caziot, J. Szpunar, R. Lobinski and M. Potin-Gautier, J. Anal. At. Spectrom., 1999, 14, 645 RSC.
- (a) S. Tagawa, Bull. Jpn. Soc. Sci. Fish., 1980, 46, 1257 Search PubMed; (b) Chem. Abstr., 1981, 94, 77823u. Search PubMed.
- Y. Inoue, Y. Date, T. Sakai, N. Shimizu, K. Yoshida, H. Chen, K. Kuroda and G. Endo, Appl. Organometal. Chem., 1999, 13, 81 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |