Application of a low power/reduced pressure helium ICP ionization source for mass spectrometric detection of organobromine compounds and derivatized organotin compounds
Joseph W. Waggonera, Lisa S. Milsteina, Mikhail Belkina, Karen L. Suttona, Joseph A. Caruso*a and Harry B. Fanninb
aDepartment of Chemistry, University of Cincinnati, Cincinnati, OH 45221-0172, USA
bDepartment of Chemistry, Murray State University, Murray, KY 42071, USA
First published on UnassignedUnassigned7th January 2000
Abstract
A low power, reduced pressure helium inductively coupled plasma source for mass spectrometry with capillary gas chromatography was applied to the detection of the organobromine species bromobenzene, 1-bromoheptane and benzyl bromide and ethylated forms of the ionic tri-substituted organotin species tributyltin chloride (TBuSnCl) and tripropyltin chloride (TPrSnCl). Total elemental information obtained with plasma source ionization was augmented with a capability to take fragment spectra that provides structural information about gas chromatographic eluents. Limits of detection for bromobenzene, 1-bromoheptane and benzyl bromide were 11, 6 and 4 pg, respectively. Molecular ions for all three organobromine compounds were obtained, as well as some characteristic alkyl chain fragments for 1-bromoheptane, resembling an electron impact mass spectrum. The ethyl, propyl and butyl substituents of ethylated TBuSnCl and TPrSnCl were clearly identified as fragments in the mass spectra. Chromatographic retention time and mass spectral matches with ethylated organotin standards were obtained for the TBuSnCl component of the certified reference material NIES-11.
Introduction
The capability of low power/reduced pressure plasma sources when coupled with mass spectrometry for providing low levels of elemental detection, and in some cases, structural information, has been documented in the literature.1–11 These experimental plasma sources could be a valuable addition to current elemental speciation methodologies for which the combination of chromatographic information with a destructive detection method, such as the conventional inductively coupled plasma, is insufficient for the identification of some chemical species.Conventional plasma source mass spectrometry, that is, a high power (>1000 W), atmospheric-pressure argon plasma (ICP), has provided enhanced sensitivity and element selectivity for organometallic analyses when compared with electron ionization mass spectrometry (EI-MS); however, the high power sources provide no molecular information. Many speciation studies thus require complementary techniques for qualitative and quantitative determinations. The new generation of plasma sources for mass spectrometry provide complementary quantitative and qualitative information with the same instrument.
In addition to demonstrating the analytical capabilities of experimental plasma sources, much research has focused on making such sources more cost effective, and thus potentially a viable addition to analytical methods. Jerrell and co-workers12 reported the use of a novel low power, reduced pressure (LP/RP) helium ICP generator with optical emission spectrometry (OES) as a detector for gas chromatography in the analysis of organohalogens. This low cost, “in-house" manufactured plasma generator, based on a design introduced by May and May for optogalvanic spectrometry,13 was assembled from readily available parts for less than 100.
A recent publication from this research group2 described the development of Jerrell's LP/RP-ICP source for elemental mass spectrometry where tetra-substituted organotin compounds were separated by gas chromatography and detected at low picogram limits of detection with a quadrupole mass spectrometer. In addition, characteristic mass spectra, resembling those of EI-MS, were obtained with this system.
The focus of this paper is to demonstrate the versatility of the aforementioned GC-LP/RP-He-ICP-MS system,2 by extending the method to (1) volatile organobromine analytes as a representative of organohalogen speciation and (2) a certified reference material (CRM) containing tri-substituted organotin species. The ability to detect organohalogens at low levels with this experimental plasma system could be applicable to the analysis of halogenated pesticides and herbicides. Tributyltin has been a contaminant of concern for some time due to its widespread use as an anti-fouling agent in boat bottom paints and the subsequent detrimental effect on marine organisms. Tributyltin can be fatal to marine life at low part-per-billion levels.
The choice of organobromine analytes and derivatized organotin compounds establishes a comparative basis with previously reported LP/RP-ICP-MS instrumentation and methods. Castillano and co-workers11 utilized an LP/RP-ICP source with mass spectrometry for the detection of the same organobromine compounds. This source was obtained by modifying the impedance matching network of the conventional ICP torchbox. However, a stable plasma could not be achieved below 90 W rf power with the Henry-type rf generator and the fragmentation was too extensive to obtain molecular spectra. O'Connor and co-workers5,8 modified both the plasma generator and matching network of a commercial ICP system which allowed for stable operation of a helium plasma down to 5 W rf power. On interfacing with a customized mass spectrometer, molecular spectra were obtained for both organometallics and organohalogens.
The gas chromatographic analysis of tri-substituted organotin species and CRMs utilizing various pre-column derivatization techniques with EI-MS or chemical ionization (CI)-MS14–16 and ICP-MS15,17–20 detection has been established. There has been little work reported involving analyses of CRMs with LP/RP plasma sources for application or method validation purposes.1,8 For this ionization source, the detection of tributyltin with structural information is the first application involving a real sample.
Experimental
Instrumentation
The plasma generator used in this work has been described in detail previously.2,12 The generator operates at 35 MHz and an estimated forward power of 12–15 W. A 48 turn copper wire load coil provides the necessary impedance to complete the matching network. Information regarding modifications to the circuit from May and May's13 original design, load coil dimensions, and power estimation is available elsewhere.2,12 An internally generated technical report for the construction of the generator is available on the world wide web.21A PlasmaQuad II STE quadrupole ICP mass spectrometer (Fisons, Winsford, Cheshire, UK) was used after removing and disabling the conventional torch box and generator. The first stage rotary pump of the instrument was used to maintain reduced pressure in the plasma torch cell and the expansion stage of the mass spectrometer. The plasma torch was a Pyrex™ tube, 1/4′′ od, 3/16′′ id, and 3.5′′ in length, and was sealed to a customized sampler (1 mm orifice) with epoxy resin. A schematic diagram and detailed description of the GC-LP/RP-He-ICP-MS interface is available elsewhere.2
Gas chromatography
Separation of the organobromine compounds and organotin compounds was accomplished using a Hewlett-Packard Model 5890 Series II gas chromatograph (Hewlett-Packard, Rockville, MD, USA). A DB-1701 capillary column (15 m × 0.32 mm id × 0.25 µm film thickness, J&W Scientific, Austin, TX, USA) was used for the organobromine separation. For the organotin work, a DB-5 capillary column (30 m × 0.32 mm id × 0.25 µm film thickness, J&W Scientific) was used. De-activated fused silica (0.32 mm id) was used for the pre-column and transfer line (J&W Scientific) for the organobromine separation. For the organotin work, use of a transfer line was avoided by threading the column directly into the plasma interface. Sample was introduced into a retention gap (fused silica pre-column) using a previously described6 pneumatic injection system with a 0.2 µl internal sample loop.Reagents and standards
Standard solutions of 1-bromobenzene, 1-bromoheptane (99 + % purity, Aldrich, Milwaukee, WI, USA) and benzyl bromide (99 + % purity, Fisher Scientific, Pittsburgh, PA, USA) were prepared in HPLC-grade hexane (Fisher Scientific). Standard solutions of tetramethyltin (TMT), triphenyltin chloride (TPhSnCl), tributyltin chloride (TBuSnCl) (Aldrich) and tripropyltin chloride (TPrSnCl) (Alfa Aesar, Ward Hill, MA, USA) were prepared and diluted in HPLC-grade methanol (Fisher Scientific, Fairlawn, NJ, USA) prior to derivatization. The organotin certified reference material was NIES-11 CRM, Fish Tissue, with certified concentrations of TBuSnCl and TPhSnCl (National Institute for Environmental Studies, Japan Environmental Agency, Onogawa, Tsukuba, Ibaraki, Japan). A 1 M sodium acetate solution (Fisher Scientific) was prepared in 18 MΩ distilled, de-ionized water (Sybon Barnstead, Boston, MA, USA). Glacial acetic acid (Fisher Scientific) was used to adjust the sodium acetate solution to pH 5. Drierite™ (Alltech Associates, Deerfield, IL, USA) was used to dry the organic layer of the extraction. Tetramethylammonium hydroxide (TMAOH) (Alfa Aesar) and HPLC-grade isooctane (Aldrich) were used for extraction. The derivatizing agent was sodium tetraethylborate (NaBEt4) (Strem Chemicals, Newburyport, MA, USA). The plasma and carrier gas was standard-grade helium (99.9% purity, Wright Brothers, Dayton, OH, USA).Microwave extraction and derivatization of organotin compounds
A procedure described by Rodriguez and co-workers22 was followed for the extraction and ethylation of the organotin chlorides found in NIES-11 CRM as well as the TPrSnCl spike. A sample amount of 0.2–0.4 g of NIES-11 CRM was added to 15 ml of TMAOH. A 100 µl volume of 10 mg kg−1 TPrSnCl standard was also added and the mixture was heated for 3 min at 120°C in the MES 1000 closed-vessel microwave extraction system (CEM, Matthews, NC, USA). After the sample vessel had been allowed to cool, 3.8 ml of glacial acetic acid, 5 ml of sodium acetate buffer (pH 5), 1 ml of isooctane, and 2 ml of ≈1% w/w NaBEt4 were added. The vessel was exposed to microwave energy at 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C for a further 2 min. The organic layer was extracted with a separating funnel, dried with Drierite™ and filtered through 0.2 µm pore size nylon syringe filters (Nalgene, Rochester, NY, USA).
°C for a further 2 min. The organic layer was extracted with a separating funnel, dried with Drierite™ and filtered through 0.2 µm pore size nylon syringe filters (Nalgene, Rochester, NY, USA).The organotin standards were ethylated in a similar manner. Single component and mixtures of 100 µl amounts of 100 mg ml−1 TPhSnCl, TBuSnCl and TPrSnCl standards were pipetted into a separatiory funnel along with 2 ml of a 1% w/w solution of NaBEt4, 5 ml of sodium acetate buffer solution (pH 5.0) and 1 ml of isooctane. The mixture was shaken for 5 min and the organic layer was extracted, dried with Drierite™, diluted with isooctane to obtain a 10 mg kg−1 standard and filtered through nylon syringe filters.
Data acquisition
The ion lenses were tuned and the capillary position in the plasma was optimized with a constant 79Br+ or 120Sn+ signal by sampling the headspace from a 10 mg kg−1 bromobenzene or TMT solution through a portion of de-activated fused silica capillary. The plasma gas flow/expansion stage pressure was optimized for maximum signal-to-background ratio with chromatographic injections of 10 mg kg−1 TMT or bromobenzene. Typical operating parameters are listed in Table 1. Data for the organobromine calibration graph and the TBuSnCl semi-quantification were acquired in single ion monitoring data acquisition mode (SIM) at m/z 79 and 120, respectively. Mass spectra were obtained in the scanning mode of the time-resolved data acquisition software (TRA).| a1 bar = 105 Pa. | |
|---|---|
| Incident rf power | 12–15 W |
| Plasma gas flow | 625–790 ml min−1 |
| Expansion stage pressure (operating) | 1.0–1.4 mbara |
| Intermediate stage pressure | 2.0 × 10−4 mbara |
| Analyzer stage pressure | 2.6–3.6 × 10−6 mbara |
| Residual cell pressure (expansion stage pressure) | 6.8 × 10−2 mbara |
| GC carrier gas flow | 8.0–11.0 ml min−1 |
| GC transfer line temperature | 270–300°C |
Results and discussion
Organobromine compounds
The GC-LP/RP-He-ICP mass spectra obtained for bromobenzene, 1-bromoheptane and benzyl bromide presented in Fig. 1 include characteristic fragmentation patterns and molecular ions. A distinct fragmentation pattern was observed for the aliphatic compound 1-bromoheptane in which six of the seven possible (CH2)x–Br fragments are detected as shown in Fig. 1(b). Fig. 1(d) is included as an example of the similarity between the LP/RP-He-ICP mass spectra and EI mass spectra. The most abundant (CH2)x–Br mass fragment obtained from this LP/RP-He-ICP mass spectrum occurs at m/z 135 which is also the case with the EI mass spectrum23 of 1-bromoheptane. Minimal mass fragmentation information is available for the aromatic organobromine compounds, which is also the case with EI-MS. A direct match of the LP/RP-He-ICP spectra with established EI mass spectral libraries has not been made at this time. The possible fragment assignments are compiled in Table 2.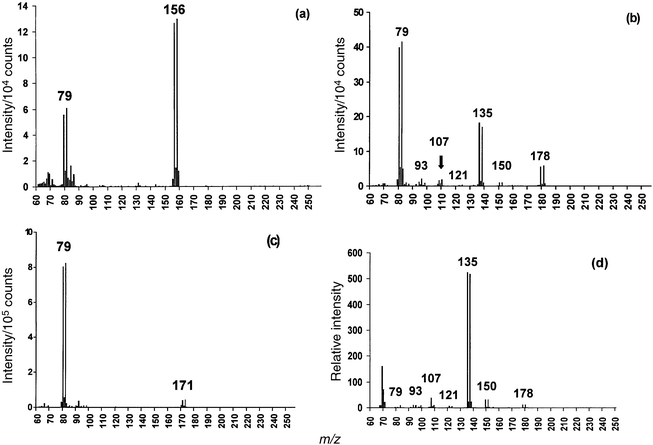 | ||
| Fig. 1 GC-LP/RP-He-ICP mass spectra of organobromine species; 26 ng injection (as Br); TRA data acquisition mode. (a) Bromobenzene. (b) 1-Bromoheptane. (c) Benzyl bromide. (d) EI mass spectra for 1-bromoheptane.23 | ||
| Fragment m/z | Fragment assignment | Compound |
|---|---|---|
| aMe = methyl; Eth = ethyl; Pr = propyl; Bu = butyl. bMolecular ion. cPossible ethylated degradation product of TPrSnCl. dPossible recombination products. | ||
| Organobromine compounds— | ||
| 79 | 79Br+ | All |
| 93 | CH2–79Br+ | 1-Bromoheptane |
| 107 | (CH2)2–79Br+ | 1-Bromoheptane |
| 121 | (CH2)3–79Br+ | 1-Bromoheptane |
| 135 | (CH2)4–79Br+ | 1-Bromoheptane |
| 150 | (CH2)5–79BrH+ | 1-Bromoheptane |
| 156 | (C6H5)–79Br+b | Bromobenzene |
| 171 | (C6H5)–CH3–79Br+b | Benzyl bromide |
| 178 | (CH2)7–79Br+b | 1-Bromoheptane |
| Organotin compounds— | ||
| 120 | 120Sn+ | All |
| 135 | (Me)–120Sn+ | All |
| 149 | (Eth)–120Sn+ | All |
| 163 | (Pr)–120Sn+ | TPrSnCl derivative |
| 177 | (Bu)–120Sn+ | TBuSnCl derivative |
| 178 | (Eth)2–120Sn+ c | TPrSnCl derivative |
| 193 | (Eth)2–Me–120Sn+d | All |
| 207 | (Eth)3–120Sn+ d | All |
| 235 | (Eth)–(Pr)2–120Sn+ | TPrSnCl derivative |
The molecular ions obtained with this source were less intense than that reported by O'Connor and co-workers8 for dibromobenzene, in which, by adding isobutane to the plasma gas, a limit of detection (LOD) of 229 pg was achieved with SIM at the molecular ion mass-to-charge ratio (m/z 236) and 76 pg with SIM at the elemental mass (m/z 81). In comparison, the LODs for organobromine compounds using the source described in this paper were 4–11 pg obtained at the elemental mass (m/z 79) and calibration could not be achieved with SIM at any of the molecular ion masses. While the source utilized by O'Connor and co-workers operated at a slightly lower rf power, it was proposed that the isobutane reduced the ionization energy in the plasma to the point where molecular ions could be obtained in high abundance.5
A representative SIM chromatogram for the organobromine separation is shown in Fig. 2. Retention times were reproducible with relative standard deviations (%RSD) less than 0.6%. Substantial peak tailing was observed and the baseline progressively decreased during the separation. These problems are most likely a memory effect attributable to the choice of stationary phase (DB-1701) and column bleed. High levels of bromine were left on the column from prior use and each successive injection and were flushed off by the solvent with each subsequent injection. Quantitative figures of merit were calculated after first subtracting the background area from each peak. The background area was determined at the front of the peak by integrating over a time interval equivalent to the width of each analyte peak.
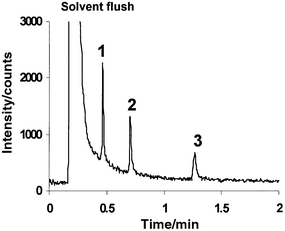 | ||
| Fig. 2 GC-LP/RP-He-ICP-MS chromatogram, SIM data acquisition mode, m/z 79; 26 pg injection (as Br). (1) Bromobenzene, (2) 1-bromoheptane and (3) benzyl bromide. | ||
The figures of merit for the detection of the species (as Br) are compiled in Table 3. The LODs (3σ/slope) were limited by the elevated baseline. Bromobenzene has the largest peak height of the three analytes, but the poorest LOD, resulting from the substantial background area subtracted and the high standard deviation of the background. The figures of merit achieved with this experimental set-up (Table 3) are comparable to those of a previous GC-LP/RP-He-ICP-MS effort from Castillano and co-workers,11 in which LODs of 16 and 7.6 pg were achieved for bromobenzene and benzyl bromide, respectively, utilizing a 100 W plasma source. This source provides comparable sensitivity for elemental detection at a much lower rf power, therefore allowing for molecular spectra capability. The combination of low picogram LOD, retention time matches and mass spectra with molecular ions, could allow for low level detection and identification of these organobromine species in an unknown sample.
| Bromobenzene | 1-Bromoheptane | Benzyl bromide | |
|---|---|---|---|
| aIntensity values background-corrected. bCalculations based on peak integrals. cLOD calculated as 3σ/slope of calibration graph.dBased on five replicate injections. | |||
| Linear range/decades | 2.5 | 2.5 | 2.5 |
| Slope/counts pg−1a,b | 152 | 199 | 151 |
| R2 value | 0.9994 | 0.9997 | 0.9994 |
| Log-log slope | 0.9448 | 0.9565 | 0.9517 |
| Limit of detectiona,b,c/pg | 11 | 6.4 | 4.2 |
| RSDd (%) | 3 | 5 | 7 |
Derivatization, extraction and detection of organotin compounds in NIES-11 CRM
Retention time matches were obtained between ethylated TBuSnCl and TPrSnCl standards and components in the spiked NIES-11 CRM as shown in Fig. 3. The CRM was spiked with a known amount of TPrSnCl (10 mg kg−1) to monitor the ethylation process; however, the derivatization efficiency was not characterized sufficiently to use TPrSnCl as an internal standard.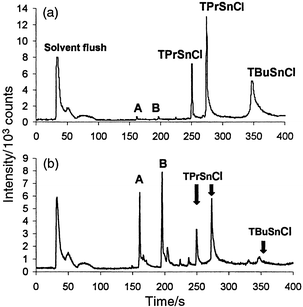 | ||
| Fig. 3 GC-LP/RP-He-ICP-MS chromatograms for ethylated organotin standards and NIES-11 CRM; SIM data acquisition mode, m/z 120; A,B unidentified. (a) TBuSnCl and TPrSnCl standard mix, 1.6 ng (as tin). (b) TPrSnCl spiked (1.6 ng as tin) NIES-11 CRM. | ||
The multiple peaks obtained for the ethylated TPrSnCl spike and other unidentified tin peaks may be attributed to the formation of degradation products at various steps of the sample preparation and analysis or from isomeric impurity in the starting reagents. Two reproducible peaks were initially attributed to TPrSnCl from retention time matching of the spiked ethylated fish sample with a single component ethylated TPrSnCl standard. Two unidentified yet reproducible small peaks (A and B in Fig. 3) were also observed in the TPrSnCl standard chromatogram and were initially considered to be minor degradation products or possibly just contaminants. However, much more intense peaks with matching retention times were observed in the NIES-11 CRM chromatogram [Fig. 3(b)]. From this observation, it appears that there could be some enhanced degradation of the TPrSnCl derivative due to the microwave extraction process similar to that reported by Rodriguez and co-workers22 for diphenyltin chloride (DPhSnCl). A more thorough study will be required to confirm this possibility. NIES report 1.3 ± 0.1 mg kg−1 tributyltin (as chloride) for NIES-11 and do not report specifically for tripropyltin. They indicate 2.4 ± 0.1 mg kg−1 as total tin for the sample.
Triphenyltin chloride (TPhSnCl) was not detected as a standard or identified as a component in NIES-11 CRM, possibly due to incomplete ethylation or severe degradation. The chromatogram for a single component ethylated TPhSnCl standard (not shown) consisted of several small and irreproducible peaks. The ethylation process in some instances may have been slowed by degradation of the NaBEt4.
Table 4 summarizes the semi-quantitative data obtained for the spiked NIES-11 CRM. The TPrSnCl spike did give a preliminary indication of the ethylation/microwave extraction efficiency by comparison of the ethylated TPrSnCl peak areas in NIES-11 CRM and the standard. The amount of the TPrSnCl detected in the NIES-11 CRM was roughly half of what was expected, suggesting that ethylation and/or extraction was incomplete. Initially, the TPrSnCl spike was intended as an internal standard to provide a correction factor for quantitative analysis, compensating for incomplete ethylation. The relative contributions of ethylation efficiency and microwave extraction efficiency to the amount of compound detected has not been determined. Further characterization of the derivatization process and the apparent degradation of TPrSnCl will be necessary to perform a fully quantitative analysis for TBuSnCl.
| TPrSnCl (ethyl derivative) | ||||
|---|---|---|---|---|
| 1st peak | 2nd peak | Sum (average) | TBuSnCl (ethyl derivative) | |
| aIntegrated raw counts. bBased on three injections of a single ethylated mixed standard. cBased on three extractions of NIES-11 CRM. dBased on proportion between the peak areas of the sample component and a 10 mg kg−1 standard. eSum of two peaks attributed to TPrSn. fNIES report 1.3 ± 0.1 mg kg−1 as chloride for TBuSnCl for NIES-11 and 2.4 ± 0.1 mg kg−1 as total tin for the sample. Tripropyltin is not reported. | ||||
| Standard peak areaa | 38603 | 95073 | 133676 | 141578 |
| RSDbfor standard peak area (%) | 9 | 4 | (7) | 20 |
| Sample peak areaa | 27207 | 59633 | 86840 | 25441 |
| RSDc for sample peak area (%) | 16 | 12 | (14) | 8 |
| Semi-quantitative concentration/mg kg−1 (as tin)d,e,f | 6.4 | 1.8 | ||
The semi-quantitative values reported in Table 4 for TBuSnCl and the TPrSnCl spike in NIES-11 CRM are based on a proportion between the CRM sample peak area and that of a 10 mg kg−1 standard. The high semi-quantitative value reported relative to the certified value may be attributed to differences in the ethylation procedure between the standard and the sample. The sample (NIES-11 CRM) was exposed to microwave energy after addition of the ethylation reagent. The TBuSnCl and TPrSnCl standards were not exposed to microwave energy during ethylation.
GC-LP/RP-He-ICP mass spectra of organotin derivatives
Characteristic mass spectra were obtained for the ethylated forms of TBuSnCl and TPrSnCl. A spectral match was obtained for the chromatographic peaks in the standard and in NIES-11 CRM sample as shown in Fig. 4(a) and (b). In addition, two chromatographic peaks initially attributed to the TPrSnCl analyte via retention time matching were shown to have characteristic propyl fragments in the mass spectra [Fig. 4(c)–(e)].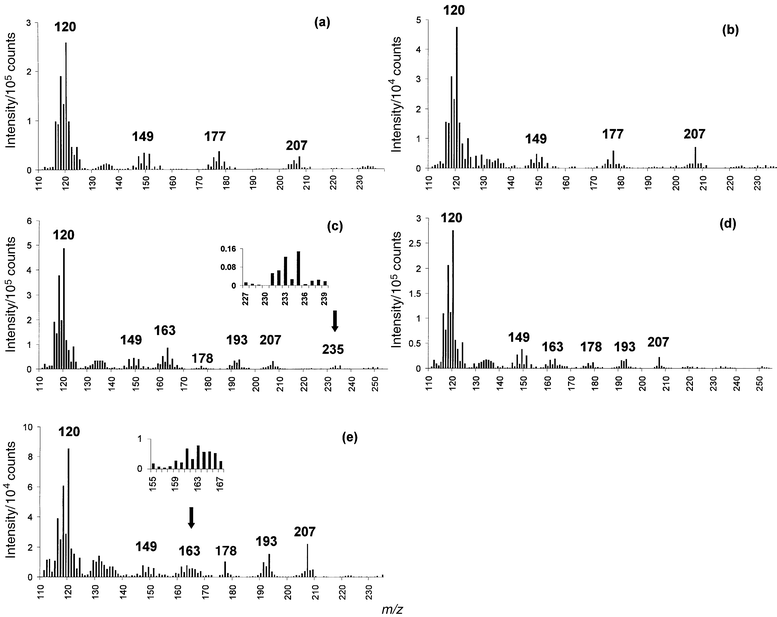 | ||
| Fig. 4 GC-LP/RP-He-ICP mass spectra for ethylated organotin compounds, TRA data acquisition mode. (a) TBuSnCl standard; 16 ng injection (as tin). (b) TBuSnCl in NIES-11 CRM. (c) TPrSnCl standard, peak 2; 16 ng injection (as tin). (d) TPrSnCl standard, peak 1; 16 ng injection (as tin). (e) TPrSnCl spike in NIES-11 CRM, peak 2. | ||
Mass spectra of derivatized compounds are difficult to interpret because the chemical form of the target analyte has been changed; however, there are some characteristic fragments that can help differentiate between compounds with different organic substituents. These fragments and possible fragment assignments are summarized in Table 2. The ethylation of these compounds is confirmed by the presence of a fragment at m/z 149. For ethylated TBuSnCl, a characteristic structural fragment is identified at m/z 177. For ethylated TPrSnCl, propyl fragments are identified at m/z 163 and 235. Some fragments in these spectra can be attributed to significant memory effects from previous injections. Other unexplained fragments may be attributed to excessive fragmentation and recombination in the plasma, in particular, the fragments at m/z 193 and 207.
The fragment assignments for m/z 193 and 207 as recombinations of ethyl group fragments are tentative, but sensible, since each analyte studied was ethylated. Such ethyl recombination products for TBuSn are atypical of EI-MS.14 While the similarities between LP/RP-He-ICP and EI mass spectra are at times striking, it is suggested by O'Connor and co-workers that there are numerous processes occurring simultaneously that contribute to the observed mass fragmentation.5
Molecular mass spectra were obtained for the unidentified peaks (labeled A,B) in Fig. 3(a) and (b), but no discernible structural fragments were observed that could help identify these components as specific alkyl or aryl tin species. The fact that these unknown components in the NIES-11 CRM fish sample are clearly tin compounds (from SIM at m/z 120) that lack structural characteristics may suggest severe degradation resulting from metabolic processes.
Comparison of the mass spectra in Fig. 4(c) and (d) for the two peaks attributed to TPrSnCl in Fig. 3(a) and (b) illustrates the value of augmenting chromatographic information with structural information in elemental speciation studies. One possible explanation for the appearance of two peaks with propyl substituents may be ethylation of a degradation product, possibly dipropyltin chloride, which would explain the higher occurrence of ethyl fragments relative to propyl fragments in the mass spectrum of the first peak. It seems reasonable that a compound with a higher degree of ethylation would exhibit more intense ethyl fragments. In addition, there is a possible fragment at m/z 178 in Fig. 4(c) and (d) which could represent the diethyl fragment ion.
Conclusions
The versatility and potential speciation applicability of a novel plasma ionization source for mass spectrometry has been demonstrated. A conventional plasma mass spectrometer that provides only total elemental information has been converted at minimal expense by use of this plasma source into an elemental and molecular mass spectrometer. This combination provides conclusive elemental speciation information to chromatographic separations for both organotin and organobromine compounds.Total element information at picogram levels of detection and extensive molecular information, including molecular ions, have been obtained for organobromine species. A previously reported study for tetra-substituted organotin compounds with this source has been extended to the less volatile ionic organotin compounds. The ethylated form of tributyltin chloride was identified in NIES-11 CRM by chromatographic retention time and mass spectral matches. The mass spectral capabilities also provide information on possible degradation products. The semi-quantitative result for TBuSnCl in NIES-11 CRM indicates that the detection of TBuSnCl may be quantifiable at low mg kg−1 levels with the system described after improvement of the derivatization and extraction technique.
Future work will focus on coupling the LP/RP-ICP-MS with low-flow liquid separation techniques, which would eliminate the need for derivatization in most cases and expand the capabilities of the instrumentation to many more speciation studies.
Acknowledgements
The authors thank the CEM Corporation for loan of the microwave extraction system, C. J. Lamb and Jim Barnett for their expertise in constructing the plasma generator, Bill Brauntz of the UC Machine Shop, Paul McKenzie and Bob Vorhees of the UC Electronics Shop and the National Institute for Environmental Health Sciences for financial support under grant number ESO4908, project 5. Additional financial support was provided by the Procter and Gamble Corporation.References
- G. O'Connor, L. Ebdon and E. H. Evans, J. Anal. At. Spectrom., 1999, 14, 1303 RSC.
- J. W. Waggoner, M. Belkin, K. L. Sutton, H. B. Fannin and J. A. Caruso, J. Anal. At. Spectrom., 1998, 13, 879 RSC.
- M. Belkin, J. W. Waggoner and J. A. Caruso, Anal. Commun., 1998, 35, 281 RSC.
- C. Brede, S. Pedersen-Bjergaard, E. Lundanes and T. Greibrokk, Anal. Chem., 1998, 70, 513 CrossRef CAS.
- G. O'Connor, L. Ebdon and E. H. Evans, J. Anal. At. Spectrom., 1997, 12, 1263 RSC.
- M. Belkin, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1997, 12, 1255 RSC.
- T. Górecki, M. Belkin, J. A. Caruso and J. Pawliszyn, Anal. Commun., 1997, 34, 275 RSC.
- G. O'Connor, L. Ebdon, E. H. Evans, H. Ding, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 1151 RSC.
- X. Yan, T. Tanaka and H. Kawaguchi, Spectrochim. Acta, Part B, 1996, 51, 1345 CrossRef.
- E. H. Evans, W. Pretorious, L. Ebdon and S. Rowland, Anal. Chem., 1994, 66, 3400 CrossRef CAS.
- T. M. Castillano, J. J. Giglio, E. H. Evans and J. A. Caruso, J. Anal. At. Spectrom., 1994, 9, 1335 RSC.
- L. J. Jerrell, M. R. Dunn, J. E. Anderson and H. B. Fannin, Appl. Spectrosc., 1999, 53, 245 Search PubMed.
- R. D. May and P. H. May, Rev. Sci. Instrum., 1986, 57, 2242 CrossRef CAS.
- R. Eiden, H. F. Scholer and M. Gastner, J. Chromatogr. A, 1998, 807, 151 CrossRef CAS.
- J. Feldmann, I. Koch and W. R. Cullen, Analyst, 1998, 123, 815 RSC.
- Z. Plzak, M. Polanska and M. Suchanek, J. Chromatogr. A, 1995, 699, 241 CrossRef CAS.
- T. De Smaele, T. Moens, L. Sandra and R. Dams, Mikrochim. Acta, 1999, 130, 241 CAS.
- R. Ritema, T. De Smaele, T. Moens, A. S. De Jong and F. X. Donard, Environ. Pollut., 1998, 99, 271 CrossRef CAS.
- E. S. Garcia, J. G. Alonso and A. Sanz-Medel, J. Mass Spectrom., 1997, 32, 542 CrossRef CAS.
- A. Prangeand and E. Jantzen, J. Anal. At. Spectrom., 1995, 10, 105 RSC.
- C. J. Lamb, Internal Technical Report, Murray State University, available on request or on the world wide web—http://campus. murraystate.edu/academic/faculty/harry.fannin/CJLAMB.HTM..
- M. Rodriguez, M. Santamarina, M. H. Bollain, M. C. Jejuto and R. Cela, Spectroscopy, 1996, 13, 51 Search PubMed.
- E. StenhagenS. AbrahamssonF. W. McLafferty, Atlas of Mass Spectral Data, Wiley, New York, 1969, p. 1200. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |