Mercury speciation by capillary gas chromatography with radiofrequency hollow cathode glow discharge atomic emission detection
Néstor Guillermo Orellana Velado, Rosario Pereiro and Alfredo Sanz-Medel*
Department of Physical and Analytical Chemistry, University of Oviedo, C/Julián Clavería, 8, 33006, Oviedo, Spain. E-mail: asm@sauron.quimica.uniovi.es
First published on UnassignedUnassigned7th January 2000
Abstract
Hollow cathode (HC) glow discharge (GD) atomic emission spectrometry (AES) with a radiofrequency (rf) source was investigated as a detector in gas chromatography (GC) for elemental speciation of methylmercury, ethylmercury and inorganic mercury following a Grignard derivatisation reaction (butylmagnesium chloride). The HC design and the transfer line between the source and the GC column are described; in addition, the parameters governing the discharge (pressure, He flow rate, rf power) on the emission of the analytes were optimised. The analytical performance characteristics of this hyphenated technique were evaluated and compared with those of other more common plasmas used as atomic detectors in GC. The absolute detection limits were found to be 0.2, 0.2 and 0.3 pg for methylmercury, ethylmercury and inorganic mercury, respectively, with the variability being lower than 8% RSD at the 10 pg level. Finally, the accuracy of the proposed methodology was successfully tested by analysing two certified reference materials from the National Research Council of Canada (DORM-2 and DOLT-2).
Introduction
Information provided by total concentration determinations of a toxic element is no longer sufficient in order to evaluate its impact on the environment, its bioavailability and its actual toxicity.1 It is recognised that the identity and concentration of the different species of the trace element of interest, that is speciation information of trace elements, is urgently needed nowadays.2 For mercury toxicity assessment, in particular, speciation of inorganic and organic forms (since methylmercury is much more toxic than inorganic mercury) is now paramount.3The combination of chromatographic techniques with atomic detectors gives rise to very powerful instruments to perform speciation analysis.4 Gas chromatography (GC) is one of the preferred separation approaches owing to its good sensitivity, resolution and availability5 when coupled to atomic absorption spectrometry (AAS),3,6 microwave-induced plasma atomic emission spectrometry (MIP-AES)7,8 and, more recently, to inductively coupled plasma mass spectrometry (ICP-MS).9,10
Glow discharges (GDs) are commonly used for direct solid elemental analysis in different fields of atomic spectrometry,11–13 but particular interest is being devoted to their use for in-depth profile analysis of layered materials.14,15 Although direct current (dc) GDs have traditionally been used in conductive solid analysis, in recent years radiofrequency (rf) GDs have been receiving particular attention since these latter discharges allow for direct solid analysis of both conductive materials and insulators.16,17 Hollow cathode (HC) plasmas are a particular type of GD,18 which has also been used for the determination of metals in solids18 and for the analysis of previously evaporated liquid samples. The analysis of liquid solutions using a particle beam system with HC-GD has also been described by Marcus and co-workers.19
Moreover, previous studies have also demonstrated the feasibility of using GDs for gas analyses. Winefordner and co-workers20 reported the atomic emission detection of Cl, Br, F, I and S for direct injection of organic solution (vapor) into a running dc-HC-GD-AES and Puig and Sacks21 used a dc-HC-GD-AES for determination of F, Cl and Br in gas streams. Also, Hieftje and co-workers described a gas sampling dc-GD-AES for determination of non-metals (C, F, Cl and S) in the gas phase.22
The application of rf-GD-AES to gas sample analyses, investigating the determination of non-metallic elements (C, Br, Cl and S) in discrete volumes of organic compounds thermally volatilised23 and of liquid samples after selective volatilisation by chemical means,24 has also been evaluated. Its excellent characteristics, observed for the analysis of different gas samples, fuelled further analytical investigations of rf-GD as an alternative specific detector in GC. For example, Smith and Piepmeier identified organic compounds using an oscillating GD as GC detector25 and Caruso and co-workers have studied a rf-GD-MS as detector in GC for elemental speciation studies.26–28 In a previous paper,29 we compared dc- and rf-GD-AES as GC detectors, with satisfactory results for mercury speciation using a flat cathode and introducing the analytes from the capillary column by the limiting disc29 (separating the cathode from the main body of the chamber).
The high emission intensities obtained with the use of very small cathode cavities, as in HC-GD (microcavity effect), could be useful for GC detection, since band broadening in the detector would be minimised.21 Considering this feature of HC designs, along with the good performance of rf-GD-AES for gas analysis,23,29 in this work a rf-HC-GD-AES was constructed and investigated as a specific GC detector for speciation of methylmercury, ethylmercury and inorganic mercury. After derivatisation with a Grignard reagent (butylmagnesium chloride), the derivatised mercury species are separated in a GC capillary column and Hg emissions are measured by AES at 253.6 nm. The transfer line design is described while the parameters affecting the performance of rf-HC-GD coupled with GC are discussed. Analytical figures of merit are established and compared with those previously obtained using a flat cathode-GD-AES29 or MIP-AES.30
Experimental
Instrumentation
Table 1 contains a description of the experimental set-up employed, including the chromatographic separation system as well as the atomic emission detection. Details of the dischargechamber,31 the rf power supply and the vacuum system are given elsewhere.29 The GC-GD interface (Fig. 1) was made from a stainless-steel tube (150 mm long, 10 mm od and 5 mm id) and the capillary column from the GC passed through its hollow interior. One end of the transfer line was introduced into the chromatograph oven while the other end was welded to the hollow cathode body. | ||
| Fig. 1 Diagram of the transfer line, hollow cathode and limiting disc. | ||
| Component | Description and specifications |
|---|---|
| (a) Gas chromatograph | Unicam Model 4600, split/splitless injection mode |
| Capillary column | Fused-silica column SGE, 25 m × 0.32 mm id, non-polar phase |
| Chromatographic program | Injector temperature: 240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C °C |
| Initial temperature: 40°C for 1 min | |
| Ramp: 35°C min−1 to 115°C for 1 min followed by 30°C min−1 to 200°C | |
| Injected sample volume | 1 µL |
| (b) Spectrometer: | |
| Monochromator | 1 m Jobin–Yvon HR-1000 M Czerny-Turner mount with a grating of 2400 grooves mm−1. Slit width 0.1 mm |
| Photomultiplier | R-212 Hamamatsu |
| Read out system | Jobin–Yvon Spectralink controlled by a computer |
| Recorder | Shimadzu data processor (Chromatopac C-R6A) |
| Parameter measured | Chromatographic peak height |
| Wavelength emission | 253.6 nm |
The hollow cathode (Fig. 2) consisted of a stainless-steel disc (40 mm in diameter and 35 mm thick). A central cavity of diameter 1 mm was made to produce the hollow cathode effect. The capillary column was introduced into the hollow cathode by a 1 mm id orifice and various distances between the end of the capillary column and the plasma discharge were tested, the position selected being that at which the capillary was closest to the hollow cathode cavity (1 mm from the plasma discharge). In some experiments the He discharge gas was introduced by the cathode through an inlet placed at 5 mm from one end of the cathode (see Fig. 2).
 | ||
| Fig. 2 Diagram of the transversal section of the hollow cathode. | ||
The limiting disc was the same as that used in previous studies29 and consisted of a stainless-steel disc (47 mm in diameter and 20 mm thick with a central orifice of diameter 3 mm) through which the light emitted by the plasma was transmitted to the quartz window and then focused to the slit of the monochromator by a 38.1 mm diameter lens (Model 41545 from Oriel Instrument, Stratford, CT, USA). Cathode and limiting disc were refrigerated by circulating cool water (5–10°C) through a lateral hollow of diameter 3 mm crossing from one side to the other, in which pipes were welded together to the external part of the holes (Fig. 2).
Materials and reagents
Dogfish Muscle and Liver certified reference materials (DORM-2 and DOLT-2) were obtained from the National Research Council of Canada (NRCC).The reagents and solutions used for the extraction and derivatisation of the sought mercury species were the same as those employed in previous studies.29 Standard solutions of methylmercury chloride (ICN Biochemicals, Cleveland, OH, USA), ethylmercury chloride (ICN Biochemicals) and mercury(II) chloride (Merck, Darmstadt, Germany) were prepared and stored in the dark at 4°C.
Sodium diethyldithiocarbamate (DDTC) was obtained from Merck with a purity of 99% and butylmagnesium chloride (2.0 M solution in tetrahydrofuran) was from Aldrich (Milwaukee, WI, USA).
Helium (99.999%) from Air Liquid (Oviedo, Spain) was used as ‘carrier gas' in the gas chromatograph and also as ‘make-up' gas to sustain the plasma discharge. The carrier was kept constant at 15 psi pressure during all experimental steps.
Procedures
For optimisation purposes, data were obtained from the actual separation of a mixture containing methylmercury, ethylmercury and Hg(II) standard solutions. The average of three replicates was considered for each investigated operating condition.The procedures for the extraction of mercury species from certified reference materials and for their Grignard derivatisation were essentially those of Bulska and co-workers.30
About 0.25 g of the certified reference material was weighed and treated with 150 µL of concentrated HCl to destroy the proteins from the organic material. Addition of 800 µL of saturated sodium chloride solution followed, in order to facilitate the formation of chlorides of the mercury species. Such species were extracted with 1.5 mL of 2 M sodium diethyldithiocarbamate and two portions of 2 mL of toluene. Ethylmercury chloride was used as an internal standard and hence was added to the reference materials before the addition of concentrated HCl.
It is known that when the MIP is used as a GC detector it is necessary to use a solvent venting system to avoid plasma extinction when the solvent exits the capillary column.32 In our HC-GD experimental system, the plasma was turned on once the solvent passed the capillary column and was carried out of the discharge chamber, at 2.5 min after injection into the gas chromatograph.
Results and discussion
Optimisation of rf power
The observed effect of the rf delivery power on the emission signal is shown in Fig. 3 for the three species under study. As can be seen, the maximum analyte signal was obtained around 30 W; however, the reflected power (2 or 3 W) produced overheating in the cathode and the limiting disc, thus increasing the emission background and giving rise to instabilities in the discharge. | ||
| Fig. 3 Effect of the rf power on the analytical signal of rf-HC-GD-AES. (✦) Methylmercury, (■) ethylmercury and (▲) inorganic mercury. | ||
Therefore, the rf power value selected was slightly lower, viz. 25 W (in this case the reflected power was zero). The results obtained in this experiment with rf-HC-GD seem to be different from those observed previously with a flat cathode,29 where up to 70 W can be delivered to the rf source without noticeable reflected energy.
Effect of plasma pressure
Different plasma pressures, keeping the He flow rate constant, were tested by changing the displacement volume of the vacuum pump using a vacuum valve with a handle rotary knob placed in the outlet of the rotary pump. As can be seen in Fig. 4, the maximum emission intensity for the mercury species occurred at about 25 Torr. Higher plasma pressures produced instabilities in the discharge and increased the background emission signal. A similar plot to that observed in Fig. 4 was obtained using a flat cathode for mercury speciation29 and different analytes.23 | ||
| Fig. 4 Effect of the plasma pressure on the analytical signal of rf-HC-GD-AES. (✦) Methylmercury, (■) ethylmercury and (▲) inorganic mercury. | ||
Effect of the make-up He flow rate
As shown in Fig. 5, at lower He make-up flow rates (keeping the discharge pressure constant) the signal intensity of the analyte increased, obtaining a maximum signal at about 10 mL min−1. This behaviour could be related in part to a lower dilution effect of the analytes in the plasma. However, the optimum He make-up flow rate appears at lower values than those of experiments on mercury speciation with a flat cathode29 (there 40 mL min−1 of He were selected) and at much lower values than those used for MIP-AES detection in speciation studies where flow rates between 100 and 300 mL min−1 are typically mentioned.32 | ||
| Fig. 5 Effect of He make-up flow rate on the analytical signal of rf-HC-GD-AES. (✦) Methylmercury, (■) ethylmercury and (▲) inorganic mercury. | ||
To increase the sensitivity of the system, the possibility of supporting the plasma with just the carrier gas from the GC (2.3 mL min−1 of He) was studied. A 15% increase of the analytical signal of the mercury species was observed compared with the use of plasma make-up flow rates of 10 mL min−1; however, the cathode became dirty too rapidly (after 4 or 5 injections onto the column) and so a compromise was needed. A working He make-up flow rate of 10 mL min−1 was eventually selected.
Finally, different ways of plasma make-up gas introduction into the discharge chamber (Fig. 6) were investigated: (A) through the limiting disc, (B) through the hollow cathode body, and (C) by the discharge chamber. Fig. 7 shows the results obtained for the three He introduction modes: as can be seen, similar results were observed when the He make-up flow was introduced either by the body of the discharge chamber or through the limiting disc (probably the mercury species suffer higher diffusion when they are carried to the plasma by introducing the plasma He gas through the hollow cathode, as shown in Fig. 6).
 | ||
| Fig. 6 Designs investigated for the introduction of the make-up gas. (A) Introduction through the limiting disc, (B) introduction through the cathode and (C) introduction through the discharge chamber. | ||
 | ||
| Fig. 7 Comparison of the emission rates of the mercury species when the He make-up plasma was introduced by means of the three designs under study. (A) Introduction through the limiting disc, (B) introduction through the cathode and (C) introduction through the discharge chamber. | ||
Analytical characteristics
Fig. 8 shows a typical chromatogram obtained for the injection of 1 µL of a 10 µg L−1 solution mixture of the three mercury species under investigation. The analytical performance characteristics of the hyphenated technique described here, evaluated for optimum plasma operating conditions, are given in Table 2 and are compared with the values for other plasmas proposed as GC detectors for mercury speciation studies. | ||
| Fig. 8 Typical chromatogram corresponding to the injection of a 10 µg L−1 mixture of the mercury species. 1, Methylmercury; 2, ethylmercury; and 3, inorganic mercury. | ||
Replicate analyses (n=10) of a mixed standard solution of 10 µg L−1 of the three species under study were carried out to evaluate the repeatability of the method. The results indicated that a RSD of about 5% was obtained for methylmercury and 7–8% for ethylmercury and inorganic mercury. The poorer results for the last two analytes can be attributed to heating during the chromatogram duration of the chamber discharge, cathode and limiting disc, which produced a change in the background emission and in the signals of these two mercury species.
The detection limits (DLs) were calculated as the concentration of analyte producing a peak height in the chromatogram equal to three times the standard deviation of the baseline noise. DLs were 0.2 ng mL−1 for methylmercury and ethylmercury and 0.3 ng mL−1 for inorganic mercury, which corresponds to 0.2, 0.2 and 0.3 pg of methylmercury, ethylmercury and inorganic mercury, respectively, as absolute DLs. As can be seen in Table 2, these DLs are 5–10 times better than those obtained using a flat cathode29 and are also better than those achieved by MIP-AES,30 which is the most common GC specific detector.
Certified reference materials analysis: method validation
The proposed hyphenated method using the hollow cathode discharge for Hg atomisation and excitation was applied to the determination of methylmercury in two marine reference materials, DORM-2 and DOLT-2. Each determination was averaged from five independent replicates of sample extraction and each replicate was injected in triplicate into the GC. Ethylmercury, added before the extraction of the analyte, was used as an internal standard. As can be seen in Table 3, the analytical results are in good agreement with the certified values. Typical chromatograms obtained for both fish tissues under study, after the recommended extraction and derivatisation, are shown in Fig. 9.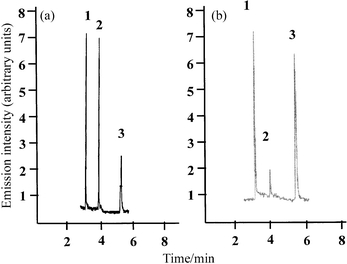 | ||
| Fig. 9 Chromatograms corresponding to the injection of an extract from the certified reference materials, after Grignard derivatisation. 1, methylmercury; 2, ethylmercury (internal standard); and 3, inorganic mercury. (a) DORM-2 and (b) DOLT-2. | ||
| Methylmercury/mg kg−1 (as Hg) | ||
|---|---|---|
| DORM-1 | DOLT-2 | |
| Obtained | 5.01 ± 0.35 | 0.620 ± 0.041 |
| Certified | 4.47 ± 0.32 | 0.693 ± 0.053 |
Conclusions
A new hyphenated system, GC-rf-HC-GD-AES, has been constructed and evaluated for mercury speciation. After optimisation of the parameters that influence mercury emission, the analytical performance observed was very satisfactory. DLs up to 5–10 times better than those encountered using flat cathode-GD-AES were obtained. As Table 2 shows, the HC-GD-AES detector is also more sensitive than the ‘reference' detection mode for GC, viz. MIP-AES.30Finally, it has to be stressed that the GC-HC-GD-AES hybrid technique proposed here presents at least one major advantage for the analysis of mercury species: the low cost of construction of the system and the relatively low consumption of He as discharge gas (10–20 times lower than that needed to operate an MIP-AES).
Acknowledgements
Financial support from DGICYT (Spain) through project number PB94-1331 is gratefully acknowledged. N.G.O.V. expresses his gratitude for the award of a scholarship from the Agencia Española de Cooperación Internacional.References
- A. Sanz-Medel, Spectrochim. Acta, Part B, 1998, 53, 197 CrossRef.
- T. D. Smaele, L. Moens, R. Dams, P. Sandra and V. Van'der Eyckens, J. Chromatogr. A, 1998, 793, 99 CrossRef.
- R. Fischer, R. Rapsomanikis and M. O. Andreae, Anal. Chem., 1993, 65, 763 CrossRef CAS.
- L. A. Ellis and D. J. Roberts, J. Chromatogr. A, 1997, 774, 3 CrossRef CAS.
- J. Szpuznar, V. O. Schmidt and R. Lobinski, J. Anal. At. Spectrom., 1996, 11, 193 RSC.
- S. Rapsomanikis and P. J. Craig, Anal. Chim. Acta, 1991, 248, 563 CrossRef CAS.
- K. J. Slatkavitz, L. D. Hoey, P. C. Uden and R. Barnes, Anal. Chem., 1985, 57, 1846 CrossRef CAS.
- M. Ceulemas, S. Slaets and F. Adams, Talanta, 1998, 46, 395 CrossRef.
- R. Lobinski and F. C. Adams, Spectrochim. Acta, Part B, 1997, 52, 1865 CrossRef.
- M. Heisterkamp, T. De Smaele, J. P. Candelone, L. Moens, R. Dams and F. Adams, J. Anal. At. Spectrom., 1997, 12, 1077 RSC.
- W. W. Harrison, C. M. Barshick, J. A. Klinger, P. H. Ratliff and Y. Mei, Anal. Chem., 1990, 62, 943A CAS.
- V. Pavski and C. L. Chakrabarti, Appl. Spectrosc., 1995, 49, 927 Search PubMed.
- M. R. Winchester and R. K. Marcus, J. Anal. At. Spectrom., 1990, 5, 575 RSC.
- J. A. C. Broekaert, Appl. Spectrosc., 1995, 49, 12A Search PubMed.
- M. Fernández, N. Bordel, R. Pereiro and A. Sanz-Medel, J. Anal. At. Spectrom., 1997, 12, 1209 RSC.
- C. Lazic and R. K. Marcus, Spectrochim. Acta, Part B, 1993, 48, 863 CrossRef.
- D. C. Duckworth and R. K. Marcus, J. Anal. At. Spectrom., 1992, 7, 711 RSC.
- S. Caroli, Prog. Anal. At. Spectrosc., 1983, 6, 253 Search PubMed.
- J. You, P. A. Depalma and K. Marcus, J. Anal. At. Spectrom., 1996, 11, 483 RSC.
- K. C. Ng, A. Ali and J. D. Winefordner, Spectrochim. Acta, Part B, 1991, 46, 309 CrossRef.
- L. Puig and R. Sacks, Appl. Spectrosc., 1989, 43, 801 Search PubMed.
- T. K. Starn, R. Pereiro and G. M. Hieftje, Appl. Spectrosc., 1993, 47, 1555 Search PubMed.
- G. Centineo, M. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Chem., 1997, 69, 3702 CrossRef CAS.
- J. Rodríguez, R. Pereiro and A. Sanz-Medel, J. Anal. At. Spectrom., 1998, 13, 911 RSC.
- D. L. Smith and E. H. Piepmeier, Anal. Chem., 1995, 67, 1084 CrossRef CAS.
- L. K. Olson, M. Belkin and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 491 RSC.
- M. Belkin, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1997, 12, 1255 RSC.
- M. Belkin, J. W. Waggoner and J. A. Caruso, Anal. Commun., 1998, 35, 281 RSC.
- N. G. Orellana Velado, R. Pereiro and A. Sanz-Medel, J. Anal. At. Spectrom., 1998, 13, 905 RSC.
- E. Bulska, D. C. Baxter and W. Frech, Anal. Chim. Acta, 1991, 249, 543 CrossRef.
- R. K. Marcus, J. Anal. At. Spectrom., 1993, 8, 935 RSC.
- E. Bulska, J. Anal. At. Spectrom., 1992, 7, 201 RSC.
| This journal is © The Royal Society of Chemistry 2000 |