Analysis of amino acids and organometallic compounds by particle beam–hollow cathode glow discharge atomic emission spectrometry
Melissa A.
Dempster
and
R. Kenneth
Marcus
*
Department of Chemistry, Howard L. Hunter Laboratory, Clemson University, Clemson, S Carolina 29634-1905, USA
First published on 27th March 2000
Abstract
An evaluation of the particle beam–hollow cathode glow discharge atomic emission spectroscopy (PB-HC-AES) system as an element-specific detector for liquid chromatography is described for aromatic amino acids and organomercury and organolead compounds. A high-efficiency thermoconcentric nebulizer is used to introduce analyte particles into a heated hollow cathode glow discharge source for subsequent vaporization, atomization and excitation. Emission responses for hydrogen and nitrogen of amino acids in flow injection mode using 200 µL injection volumes indicate detection limits of 0.13 and 3.6 ppm elemental H and N, respectively, with RSD <10% for triplicate injections over a concentration range of 10−3–10−2 M. Analysis of the response of hydrogen and nitrogen emission signal intensities to differences in amino acid stoichiometries indicates the potential of the PB-HC-AES system for the determination of empirical formulas based on H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) I/NI intensity ratios. Detection limits for metals and non-metals in organomercury compounds as determined through flow injection of 200 µL sample volumes fall in the sub-ppm range. Liquid chromatographic separations of both amino acids and organolead compounds demonstrate the feasibility of the PB-HC-AES system as a detector for liquid chromatography. Retention times for 20 µL injections of the selected component mixtures as determined by CI emission for amino acids and PbI emission for organolead compounds correlate well with the corresponding UV absorbance results.
I/NI intensity ratios. Detection limits for metals and non-metals in organomercury compounds as determined through flow injection of 200 µL sample volumes fall in the sub-ppm range. Liquid chromatographic separations of both amino acids and organolead compounds demonstrate the feasibility of the PB-HC-AES system as a detector for liquid chromatography. Retention times for 20 µL injections of the selected component mixtures as determined by CI emission for amino acids and PbI emission for organolead compounds correlate well with the corresponding UV absorbance results.
Introduction
The proper identification and speciation assignment of trace elements in environmental and biological systems is essential in studying their mobility, absorption and toxicity. While trace metals analysis has traditionally yielded only ‘total elemental concentration' information, one area where advances in analytical methodology can have a large impact is in true chemical speciation of environmental and biological samples. It is not sufficient simply to quantitate the amount of a specific metal present in a system, as the associated complex greatly affects the toxicity. This is of great environmental concern, as contamination from industrial products has resulted in the formation of a variety of organometallic species that must be characterized in order to evaluate their potential effects on the surrounding biological systems.1As regulations are refined to reflect truly the practical toxicity and chemistry of the elements in different systems, the need for methods to evaluate both chemical speciation (oxidation state) and ligand identification increases. The ideal situation for supplying analytical information is the combination of separation techniques such as gas chromatography (GC) or liquid chromatography (LC) with compatible detection modes that can produce complete compound or empirical formula information. Determinations of empirical formulas can be achieved by calculating the ratio of the responses of the individual component elements (e.g., Cu/C/H/O) if atomic spectrochemical (elemental analysis) methods are employed. Currently, the pairing of capillary GC with microwave-induced plasma atomic emission spectrometry (GC-MIP-AES) is the only commercially available option for providing this level of information.2–5 In the commercial implementation, the MIP source is optically sampled by a photodiode array for simultaneous multielement analysis, termed GC-AED (gas chromatography–atomic emission detection).6 This approach has displayed a wide scope, offering both qualitative and quantitative information on the speciation analysis of petrochemicals and pesticide residues.3,4 The ability of the GC-MIP-AES system to provide elemental analysis and empirical formula information, which is traditionally obtained by various combustion methods, is expected to have significant impact in the speciation of volatile compounds. For example, Donais et al.5 reported a solid–liquid extraction procedure with preparative gel permeation chromatography clean-up and GC-MIP-AES analysis for the quantification of methylmercury species in a variety of complex marine materials.
The development of techniques to obtain information on the identity of ligand species for non-volatile compounds that are separated by LC methods has not progressed as well as for volatile compounds. Chromatographic methods coupled with inductively coupled plasma atomic emission (ICP-AES) or mass spectrometry (ICP-MS) are currently used to determine oxidation state information;7–9 however, the presence of entrained gases in this atmospheric plasma source limits the ability to determine elements such as O, N, H and S owing to high background signals. By taking advantage of the low-pressure environment of the particle beam–hollow cathode atomic emission spectrometry (PB-HC-AES) system, analysis of these atmospheric elements is possible, allowing assignment of empirical formulas in addition to metal speciation.
The PB interface provides a means of employing spectrochemical detection for LC eluents while maintaining natural chromatographic characteristics such as retention/elution quality and solvent gradient compatibility. It has found increasing use as a valuable sample introduction device for liquid chromatography–mass spectrometry (LC-MS) because of its simplicity of operation and compatibility with a wide range of solvent polarities and flow rates.10 The PB interface accomplishes separation and enrichment of the analyte from the solvent through the processes of nebulization (aerosol formation), desolvation and momentum separation, ultimately transferring the dry analyte particles into the detection source. Primary aerosols are generated from the LC flow by use of a nebulizer in order to expedite analyte desolvation. After passing through a heated desolvation chamber, the resulting mixture of solvent vapor and analyte particles enters a two-stage momentum separator. The heavier analyte particles are transported through the interface to the detection source for subsequent vaporization excitation or ionization, while the lighter nebulizer gas and solvent vapor are skimmed and pumped away as they pass through the differential pumping stages. In addition to performing analyte enrichment through removal of solvent, differential pumping of the momentum separator also provides reduction of pressure to match the vacuum requirements of the detection source.
The glow discharge (GD) has become well known as a versatile source for direct solids elemental analysis of both conductive and non-conductive samples by AAS, AES and MS owing to its efficient atomization, excitation and ionization processes.11–13 Although the capabilities of the GD have been established as a powerful spectrochemical source for analysis of solid samples, the extension of this technique to liquid samples has found limited interest. The use of hollow cathode glow discharges to sputter atomize solution residues has been reported, where a small aliquot of liquid sample is placed in the base of the hollow cathode and dried to a solid residue, then converted into gas-phase atoms through cathodic sputtering.14,15 The implementation of the PB interface to allow continuous sample transfer to the GD source has been demonstrated in this laboratory, with sensitivities in the ppb range for metallic elements introduced through flow injection with detection by AES.16–20
The application of the PB-HC-AES technique for the analysis of non-metals was recently explored for the case of organic compounds.20 A series of aliphatic amino acids were examined on the basis of their carbon and hydrogen emission characteristics, with resulting detection limits of 320 and 24 pg for C and H, respectively. The feasibility of this method for the determination of empirical formulas was also demonstrated through comparison of measured CI/HI emission intensity ratios to the actual atom number ratios in this series of amino acids. As the capabilities of this technique for both metals and non-metals have been established, one could anticipate the possibility of speciation analysis of organometallic compounds. Further characterization of the PB-HC-AES system is presented here for the analysis of both aromatic amino acids and organometallics. In addition, implementation of LC separations was performed for organolead compounds and for a group of underivatized amino acids to evaluate the utility of this system as an element-specific detector for LC.
Experimental
Glow discharge source
The instrumentation of the PB-HC-AES source is basically the same as applied in previous studies.17 As shown in Fig. 1, the hollow cathode is mounted at the center of a stainless steel ‘thermoblock' that ensures easy access to the plasma via optical monitoring. The source vacuum port, discharge gas inlet, pressure gauge, electrical feedthroughs and heating components are also placed on the thermoblock. The entire glow discharge source is heated for more efficient sample vaporization and atomization by a pair of cartridge heaters (Model SC 2515, Scientific Instrument Services, Ringoes, NJ, USA), with the block temperature monitored by a W–Re thermocouple. The overall gas pressure of the glow discharge source (a combination of helium discharge gas and possibly other gaseous species from the particle beam interface) is monitored by a vacuum gauge (Model 912011, VRC, Pittsburgh, PA, USA). The hollow cathode discharge is powered by a Bertan (Hicksville, NY, USA) Model 915-1N supply, operating in a constant-current mode, in the range 0–100 mA.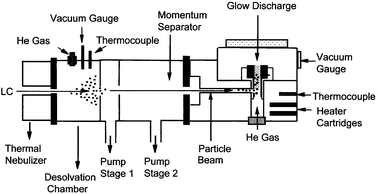 | ||
| Fig. 1 Diagrammatic representation of the PB-HC-AES apparatus. | ||
Particle beam interface
A Thermabeam (Extrel, Pittsburgh, PA, USA) LC-MS particle beam interface is employed to couple the liquid delivery system with the hollow cathode glow discharge source. The interface consists of a thermoconcentric nebulizer, a heated spray chamber and a two-stage momentum separator.10,16–18 The liquid sample is introduced via an HPLC pump into a fused-silica capillary (110 µm id) mounted within a stainless steel tube (1.6 mm od). A dc potential difference across the stainless steel tubing results in resistive heating along the tube, producing a thermal component to the aerosol formation. Helium gas flows (∼360 mL min−1) through the gap between the central fused-silica capillary and outer stainless steel tubing as a sheath gas to improve heat conduction and to break up the liquid at the capillary tip (i.e., pneumatic nebulization). The resulting fine aerosol is directed into a 35 mm diameter, 110 mm long steel spray chamber wrapped in heating tape (150–200°C). An additional 6 mm id inlet allows a supplemental flow (250 mL min−1) of He gas.18 The separation of the aerosol mixture is achieved in the subsequent two-stage momentum separator that skims out the relatively low mass nebulizer gas and solvent molecules, effectively enriching the analyte beam in the differential pumping regions. After passing through the momentum separator, the resultant beam of dry particles enters the heated (220°C) hollow cathode glow discharge volume for subsequent vaporization, atomization and excitation.
Sample preparation and solution delivery
Aqueous stock standard solutions of reagent-grade amino acids (Sigma, St. Louis, MO, USA), organomercury compounds (Aldrich, Milwaukee, WI, USA), and organolead compounds (Aldrich and Alfa Aesar, Ward Hill, MA, USA) were prepared with HPLC-grade water (Fisher Scientific, Pittsburgh, PA, USA). The solvent and analyte solutions are delivered in a continuous mode from a solvent reservoir to the nebulizer by a Waters (Milford, MA, USA) Model 600S HPLC pump. When operated in the flow injection mode, the sample is introduced to the liquid flow at through a manual Rheodyne (Cotati, CA, USA) Model 7725i sample injector with either a 20 or 200 µL sample loop volume. Separations of organolead compounds were performed by gradient elution from 40 to 90% methanol over 10 min at 1.0 mL min−1 with an ion-pair concentration (sodium 1-pentanesulfonate) of 4 mM at pH3.0 using a Waters NovaPak C18 column (150 × 3.9 mm id). Amino acids were separated with a Vydac (Hesperia, CA, USA) C18 column (250 × 4.6 mm id) using a mobile phase of 0.02% aqueous phosphoric acid–methanol (98 + 2) at 1.0 mL min−1.
Optical spectrometer and data acquisition system
The hollow cathode emission is sampled optically by use of a plano-convex fused-silica lens (45 mm diameter, 10 cm focal length) such that the image of the excitation region is focused ∼1∶1 on to the entrance slit of the monochromator. A 0.24 m Czerny–Turner spectrometer equipped with a 2400 grooves mm−1 holographic grating (Digikrom 240, CVI Laser, Albuquerque, NM, USA) is applied for optical monitoring, with the scanning range, slit width, spectral calibration and wavelength selection adjusted via the monochromator control interface. The atomic emission signals detected by a photomultiplier tube (Model R955, Hamamatsu, Bridgewater, NJ, USA) are converted into voltage signals with an analog current meter. A Macintosh IIsi computer is employed to record the output of the current meter via a National Instruments (Austin, TX, USA) NB-MIO-16X interface board. For this particular application, an X–Y recorder-type program within the National Instruments LabView 2 software environment has been developed to record the data. Finally, the digital data obtained are processed and managed in the form of Microsoft (Seattle, WA, USA) Excel files.Results and discussion
Aromatic amino acids
Previous studies of emission responses of C and H in aliphatic amino acids by PB-HC-AES demonstrated the potential of this technique for determination of empirical formulas in organic compounds.20 Emission responses obtained for aromatic amino acids are similar to those obtained for aliphatic amino acids. Analytical response curves for HI emission at 656.3 nm and NI emission at 575.3 nm were determined for triplicate 200 µL injections of DL-tryptophan at a liquid (HPLC-grade H2O) flow rate of 1.5 mL min−1 over the concentration range 10−3–10−2 M DL-tryptophan. The transient peak areas were calculated within Microsoft Excel by first subtracting the background for a time-scale analogous to that of the peak, followed by integrating the total signal from the peak starting point over a fixed peak width. Triplicate injections at each analyte concentration produce variations of RSD <10%. Response functions of y = 2 × 107x + 8948.4 for HI emission and y = 8 × 105x
− 842.4 for NI emission were obtained, with correlation coefficients of 0.9956 and 0.997, respectively. Based on these calibration data, detection limits (DL = 3sblank/m) for DL-tryptophan are calculated to be 2.2× 10−9 mol (2.2 ppm) for HI emission and 2.5× 10−8 mol (25.9 ppm) for NI emission. These amounts correspond to 0.13 ppm elemental hydrogen and 3.6 ppmelemental nitrogen. These values compare well with those given by the established method of indirect chemiluminescence detection of amino acids (2 × 10−9 mol).21 Fluorescence detection of pre-column HSAA derivatives (6.4 × 10−15 mol),22 UV–visible absorbance of pre-column BZTC and PTC derivatives (3.9 × 10−12 mol)23 and UV–visible absorbance of post-column ninhydrin complexes (1.5 × 10−11 mol)24 currently exhibit greater sensitivity than the PB-HC-AES technique; however, these methods require chemical modification steps and lack the ability to provide elemental information. These data demonstrate the capability of this system for basic elemental analysis of hydrogen and nitrogen in aromatic amino acids.
Fig. 2a and b show the response of HI and NI emission intensities for a series of equimolar solutions of some aromatic amino acids, having hydrogen and nitrogen atom numbers ranging from 9 to 12 and from 1 to 3, respectively. This group of 1 × 10−2 M DL-amino acids (proline, histidine, β-3,4-dihydroxyphenylalanine and tryptophan) display a proportional relationship between the integrated emission signal and the number of atoms in the individual molecules for both hydrogen and nitrogen. The degree of correlation for these response curves is lower than that obtained previously20 for CI and HI emission in aliphatic amino acids. This outcome may possibly be due to differences in the atomization/excitation efficiencies for the more complex aromatic structures, and could probably be overcome through the use of higher thermoblock temperatures and/or increased discharge currents.
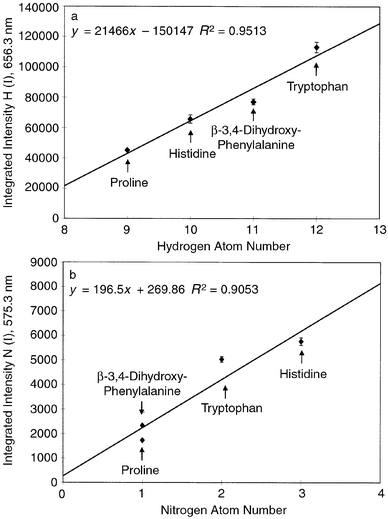 | ||
| Fig. 2
Response curves obtained for integrated H and N emission intensities as a function of number of atoms per mole of amino acid. (a) HI 656.3 nm emission; (b) NI 575.3 nm emission. Error bars represent the range of values for triplicate 200 µL injections of 10−2 M solutions. Discharge current, 40 mA; source pressure, 2.5 Torr He; nebulizer temperature, 280°C.
| ||
The potential of the PB-HC-AES technique for the determination of empirical formulas in organic compounds is illustrated in Fig. 3. This plot compares the ratio of the HI/NI integrated emission intensities to the actual ratio of those elements in the series of aromatic amino acids. The experimental HI/NI intensity values are not expected to equal the actual atom ratio densities, as each element's transition differs substantially in sensitivity. However, a linear relationship exists for these ratios with a higher degree of correlation than was achieved for the separate HI and NI emission plots, even though the data for each element were collected on different days. Clearly, as in the case of the commercial GC-AED system, the use of a diode array spectrometer, or the like, to monitor the target elements simultaneously would yield higher levels of accuracy along these lines. These results indicate the possible utility of this technique for the generation of empirical formula information.
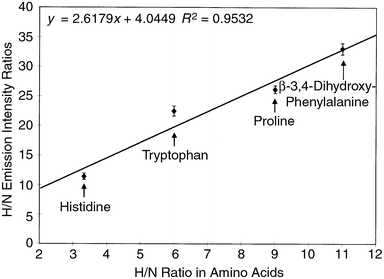 | ||
| Fig. 3
Comparison of experimentally obtained HI 656.3 nm/NI 575.3 nm emission intensity ratios to the actual atom ratios (H/N) for the range of amino acids.
| ||
Organomercury compounds
The ability of the PB-HC-AES system to determine both metals and atmospheric elements (C, H, N) provides an opportunity for the analysis of organometallic compounds. Similarly to the case for aromatic amino acids, empirical formulas based on the emission ratios of the elements present in a compound can be determined and used to identify the ligand associated with a metal. This information is important both biologically and environmentally, as the chemical form affects the toxicity and reactivity of these organometallics. Table 1 shows calibration data for various elements present in 1 × 10−4–1 × 10−2 M solutions of two organomercury compounds, thimerosal (C9H9HgO2SNa) and merbromin (C20H8Br2HgO6Na2). The chemical structures of these compounds are shown in Fig. 4. These data illustrate the capability of the PB-HC-AES technique for the determination of both metals and non-metals in organometallic compounds. Each element analyzed produced linear calibration results with good regression coefficients and sensitivity. The values for Hg detection compare well with those given by the established method of UV detection of mercury compounds separated by ion-pair HPLC (0.14–0.74 ppm Hg).25 ICP-AES of preconcentrated mercury also produces comparable detection limits of 0.1226 and 0.004 ppm Hg.27 However, the atmospheric nature of the ICP limits the analysis of non-metallic elements such as C, H and N, which can be determined using the PB-HC-AES system by taking advantage of the low-pressure environment.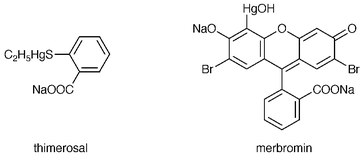 | ||
| Fig. 4 Structures of the organomercury compounds thimerosol and merbromin. | ||
| Analyte | Wavelength/nm | Response function | r 2 | Detection limit |
|---|---|---|---|---|
| Hg in thimerosal | 435.8 | y = 9 × 107x − 17964 | 0.9979 | 2.9 × 10−11 mol thimerosal (0.03 ppm Hg) |
| Hg in merbromin | 435.8 | y = 6 × 107x − 7661.8 | 0.9995 | 4.1 × 10−11 mol merbromin (0.04 ppm Hg) |
| C in thimerosal | 538.0 | y = 3.2 × 105x − 160.28 | 0.9834 | 3.5 × 10−8 mol thimerosal (19 ppm C) |
| C in merbromin | 538.0 | y = 2.2 × 105x + 45.316 | 0.9934 | 5.2 × 10−8 mol merbromin (63 ppm C) |
| Na in thimerosal | 589.0 | y = 1 × 108x − 10455 | 0.9943 | 1.4 × 10−10 mol thimerosal (0.02 ppm Na) |
| Br in merbromin | 614.9 | y = 8 × 106x − 1022.4 | 0.9997 | 1.3 × 10−10 mol merbromin (0.1 ppm Br) |
| H in thimerosal | 656.3 | y = 1 × 107x + 8977.9 | 0.9877 | 2.2 × 10−9 mol thimerosal (0.1 ppm H) |
Ideally, the emission response curves for each element should be proportional to the number of atoms in the respective compounds. This is the case for HgI emission, where a similar slope is produced for equimolar concentration ranges of thimerosal and merbromin, each of which contains one Hg atom. Because of the difference in carbon atom numbers for these two compounds (nine for thimerosal, 20 for merbromin), a larger (∼2×) slope would be expected for the CI emission response curve for merbromin. However, the data are almost the reverse of this. This may be due to incomplete atomization and excitation of the much larger merbromin molecule within the glow discharge. As Fig. 4 illustrates, merbromin consists of a fused aromatic ring which may not completely dissociate within the plasma. This would, of course, affect the CI response, whereas the Hg, H, Na and Br are on the periphery of the ring system and are easily dissociated. The disparity between the two CI responses could possibly be remedied through the utilization of a higher discharge current or hollow cathode temperature. A similarly fused system to merbromin is not available, but investigations of the role of chemical structure will be investigated in future studies. In any case, the sensitivity and range of potential analyte species depicted in Table 1 are very promising, particularly considering the relatively simple emission spectrometer system employed here.
Separation of organolead compounds
The ultimate goal of this project is development of the PB-HC-AES system as a practical element-specific detector for LC. The previously described studies illustrate the analytical capabilities of this technique for single compounds in the flow injection mode, but implementation of chromatographic separations has not been demonstrated thus far. A separation of organolead compounds with ICP-MS detection was adapted from Al-Rashdan et al.28 as a test-case separation for the PB-HC-AES system. In that work, as with any other ICP-based method, only the central metal was monitored (i.e., 208Pb+) as each compound eluted. A Waters NovaPak C18 column was employed for separation of 150 ppm each of lead nitrate, triethyllead chloride and triphenyllead chloride. The separation was performed using gradient elution from 40 to 90% methanol over 10 min at 1.0 mL min−1, with an ion-pair concentration (sodium 1-pentanesulfonate) of 4 mM at pH3.0. Fig. 5(a) shows the resulting chromatogram for a 20 µL injection with UV absorbance detection at 254 nm. The corresponding separation with PB-HC-AES detection at the PbI 405.8 nm emission line is displayed in Fig. 5(b). The retention times in each chromatogram are very similar, with the peaks detected by AES just slightly later than that for UV owing to the time required to traverse the PB interface. A small shift in baseline occurs as the gradient progresses to a higher percentage of methanol, but travel through the PB does not appear to contribute to peak broadening or tailing. Detection limits for each of the compounds are of the order of tens of nanograms (single ppm) of Pb.
![HPLC separation of 150 ppm lead nitrate [Pb(NO3)2], triethyllead (TEL) and triphenyllead (TPhL). (a) UV detection at 254 nm; (b) PB-HC-AES detection at the Pb I 405.8 nm emission line. Waters NovaPak C18 column (150 × 3.9 mm id); 4 mM sodium 1-pentanesulfonate at pH 3.0; gradient elution from 40 to 90% methanol over 10 min; flow rate, 1 mL min−1; injection volume, 20 µL.](/image/article/2000/JA/a905812e/a905812e-f5.gif) | ||
| Fig. 5
HPLC separation of 150 ppm lead nitrate [Pb(NO3)2], triethyllead (TEL) and triphenyllead (TPhL). (a) UV detection at 254 nm; (b) PB-HC-AES detection at the PbI 405.8 nm emission line. Waters NovaPak C18 column (150 × 3.9 mm id); 4 mM sodium 1-pentanesulfonate at pH3.0; gradient elution from 40 to 90% methanol over 10 min; flow rate, 1 mL min−1; injection volume, 20 µL.
| ||
Attempts to monitor CI or HI emission for the eluting compounds were unsuccessful owing to the high background levels for these elements, as the dried salt particles of the ion-pairing agent (sodium 1-pentanesulfonate) are continuously delivered to the glow discharge. In short, the addition of involatile ion-pairing or buffer species in the LC eluent (as opposed to neat separations) effectively adds non-analyte species to the PB. This is clearly an issue that must be addressed in future work. However, this separation demonstrates the potential of the PB-HC-AES technique as a detector for LC.
Separation of amino acids
To illustrate the capabilities for detection of organic compounds, an isocratic separation of underivatized amino acids was adapted from Hancock et al.29 A Vydac C18 column was used to separate a mixture of 1 × 10−2 M each of two aromatic amino acids (histidine and β-3,4-dihydroxyphenylalanine) and two aliphatic amino acids (valine and methionine). A mobile phase of 0.02% phosphoric acid–methanol (98 + 2) at 1.0 mL min−1 was employed, which allows for the separation of the underivatized amino acids with UV detection at 190 nm. The resulting chromatograms for 20µL injections of the solutions with UV and AES detection are shown in Fig. 6(a) and (b), respectively. The retention times for the peaks detected at the CI 583.0 nm emission line correlate well with those determined by UV absorbance. The emission peak for valine in Fig. 6(b) appears to be split, but a visual examination of the background noise suggests that this is probably due to background fluctuations in the plasma rather than a chromatographic effect [which is not seen in Fig. 6(a)]. Ideally, the emission peak intensities should correspond to the number of carbon atoms present in each amino acid, as equimolar concentrations were injected. However, peak areas for histidine (six C atoms) and β-3,4-dihydroxyphenylalanine (nine C atoms) are not much different from those for valine and methionine (five C atoms each). As mentioned previously, this effect may be due to differences in the atomization/excitation efficiencies for the aromatic and aliphatic structures, as the response for the aromatic compounds is less than that for the aliphatic molecules. The use of higher vaporization temperatures will be investigated through replacement of the currently used Teflon insulator for the hollow cathode with boron nitride, as the practical operating temperature is currently limited by the melting of Teflon at ∼280°C.
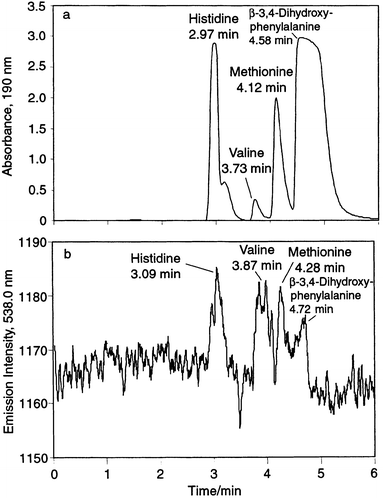 | ||
| Fig. 6
HPLC separation of 10−2 M histidine, valine, methionine and β-3,4-dihydroxyphenylalanine. (a) UV detection at 190 nm; (b) PB-HC-AES detection at the CI 583.0 1nm emission line. Vydac C18 column (250 × 4.6 mm id); 0.02% phosphoric acid–methanol (98 + 2); isocratic elution; flow rate, 1 mL min−1; injection volume, 20 µL.
| ||
Conclusions
The potential of the PB-HC-AES system as an element-specific detector for LC has been demonstrated. Studies of HI and NI emission in aromatic amino acids suggest a capability for empirical formula determinations. Encouraging response curve statistics were obtained for a variety of elements in organomercury compounds, indicating the utility of this technique for the analysis of both metals and their associated ligand species. Chromatographic separations display good correlation between UV and PB-HC-AES detection modes. Future work will focus on the optimization of parameters such as wall temperature and discharge current to overcome differences in the atomization/excitation efficiencies of various molecular structures, and also the use of more volatile buffers to limit the background produced by mobile phase modifiers transported across the PB interface. A photodiode array will also be implemented to allow the simultaneous monitoring of multiple elements. Although further work is required to optimize this technique for use with a variety of chromatographic eluents, these preliminary studies demonstrate the potential of the PB-HC-AES system as a practical element- and ligand-specific detector for LC.
Acknowledgements
Financial support from the National Science Foundation under Grant CHE-9767227 and the donation of the Thermabeam nebulizer and particle beam interface by Extrel Corporation are greatly appreciated.References
- Organometallic Compounds in the Environment: Principles and Reactions, ed. P. J. Craig, John Wiley and Sons, New York, 1986, pp. 1–32. Search PubMed.
- J. Szpunar-Lobinska, M. Ceulemans, R. Lobinski and F. C. Adams, Anal. Chim. Acta, 1993, 278, 99 CrossRef CAS.
- J. A. Seeley, Y. Zeng, P. C. Uden, T. I. Eglington and I. Ericson, J. Anal. At. Spectrom., 1992, 7, 979 RSC.
- K.-C. Ting and P. Kho, J. Assoc. Off. Anal. Chem., 1991, 74, 991 Search PubMed.
- M. K. Donais, P. C. Uden, M. M. Schantz and S. A. Wise, Anal. Chem., 1996, 68, 3859 CrossRef.
- HP G2350A Atomic Emission Detector For Gas Chromatography, Hewlett-Packard, Palo Alto, CA, 1995. Search PubMed.
- D. J. Gerth and P. N. Kelliher, in Element-Specific Chromatographic Detection by Atomic Emission Spectroscopy, ed. P. C. Uden, American Chemical Society, Washington, DC, 1992, ch. 16, pp. 275–287. Search PubMed.
- N. P. Vela, L. K. Olson and J. A. Caruso, Anal. Chem., 1993, 65, 585A CAS.
- S. J. Hill, M. J. Bloxham and P. J. Worsfold, J. Anal. At. Spectrom., 1993, 8, 499 RSC.
- C. S. Creaser and J. W. Stygall, Analyst, 1993, 118, 1467 RSC.
- W. W. Harrison, C. M. Barshick, J. A. Klingler, P. A. Ratliff and Y. Mei, Anal. Chem., 1990, 62, 943A CAS.
- R. K. Marcus, Glow Discharge Spectroscopies, Plenum Press, New York, 1993. Search PubMed.
- R. K. Marcus, T. R. Harville, Y. Mei and C. R. Shick, Anal. Chem., 1994, 66, 902A CAS.
- W. W. Harrison and N. J. Prakash, Anal. Chim. Acta, 1970, 49, 151 CrossRef CAS.
- F. Chen and J. C. Williams, Anal. Chem., 1990, 62, 489 CrossRef CAS.
- C. M. Strange and R. K. Marcus, Spectrochim. Acta, Part B, 1991, 46, 517 CrossRef.
- J. You, J. C. Fanning and R. K. Marcus, Anal. Chem., 1994, 66, 3916 CrossRef CAS.
- J. You, P. A. DePalma, Jr. and R. K. Marcus, J. Anal. At. Spectrom., 1996, 11, 483 RSC.
- J. You, M. Dempster and R. K. Marcus, J. Anal. At. Spectrom., 1997, 12, 807 RSC.
- J. You, M. Dempster and R. K. Marcus, Anal. Chem., 1997, 69, 3419 CrossRef CAS.
- P. J. Koerner and T. A. Nieman, Mikrochim. Acta, 1987, 11, 79.
- J. You, X. Fan, Q. Zhu and Y. Su, Anal. Chim. Acta, 1998, 367, 69 CrossRef CAS.
- K.-L. Woo and Y.-K. Ahan, J. Chromatogr., 1996, 740, 41 CrossRef.
- Biochrom 20 Amino Acid Analyser, Pharmacia Biotech, Cambridge, 1998. Search PubMed.
- Y.-S. Ho and P. C. Uden, J. Chromatogr., 1994, 688, 107 CrossRef CAS.
- S. Arpadjan, L. Vuchkova and E. Kostadinova, Analyst, 1997, 122, 243 RSC.
- P. C. Rudner, A. G. de Torres, J. M. C. Pavon and F. S. Rojas, Talanta, 1998, 46, 1095 CrossRef CAS.
- A. Al-Rashdan, N. P. Vela, J. A. Caruso and D. T. Heitkemper, J. Anal. At. Spectrom., 1992, 7, 551 RSC.
- W. S. Hancock, C. A. Bishop and M. T. W. Hearn, Anal. Biochem., 1979, 92, 170 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |