Capillary gas chromatography coupled with negative ionization microplasma mass spectrometry for halogen-selective detection
Cato Brede*a, Stig Pedersen-Bjergaardb, Elsa Lundanesa and Tyge Greibrokka
aDepartment of Chemistry, University of Oslo, P.O. Box 1033, Blindern, 0315, Oslo, Norway. E-mail: cato.brede@kjemi.uio.no
bSchool of Pharmacy, University of Oslo, P.O. Box 1068, Blindern, 0316, Oslo, Norway
First published on UnassignedUnassigned7th January 2000
Abstract
Gas chromatography was coupled with plasma mass spectrometry with a microplasma ion source for negative ion detection. The ion source, which was kept inside the high vacuum chamber of the mass spectrometer, was a rigid fused silica capillary tube containing a capacitively coupled radiofrequency helium plasma. This made the setup quite simple: eliminating the sampler–skimmer pressure-reducing interface traditionally used in plasma mass spectrometry. The present study describes the utilization of halogen-selective negative ion detection. A high selectivity of fluorine to hydrocarbon compounds (3 × 103), and a highly sensitive detection for F, Cl, Br, and I (0.13–12 pg s−1), were obtained. The mechanisms of negative ion formation and breakdown were discussed in conjunction with the results that were achieved.
Introduction
Capillary gas chromatography (GC) is a commonly used technique for the separation of volatile organic compounds in complex samples. The analytes are carried through long open tubular capillary columns fast, without significant band broadening, due to the low viscosity and high diffusion of the mobile phase gas. Thus, highly efficient separations are obtained, and if standard compounds are available, compound determination can be performed by using the retention times for identification. However, difficult samples might occur, in which the amount or number of the different matrix compounds exceeds the limit for obtaining sufficient chromatographic resolution. In such cases, selective detection might ease the sample preparation, which can be difficult when the analyte is present at trace levels. In order to achieve increased detection selectivity, the analyte must have somewhat different chemical properties than the matrix compounds, and this difference must be possible to utilize instrumentally. Therefore, many physical and chemical features have been explored in order to achieve selective and sensitive chromatographic detection. One such selective GC detector is the electron capture detector (ECD), which utilizes the electron affinity of analytes for highly sensitive detection, and has therefore found widespread use for the determination of halogenated compounds. Two other examples are the nitrogen phosphorus detector (NPD) and the S- and P-selective flame photometric detector (FPD). However, all of the detectors mentioned above have a limited versatility compared to the popular bench-top mass spectrometer (MS). In the electron ionization (EI) mode, the MS provides specific fragmentation mass spectra which can be used for compound identification. In the chemical ionization (CI) mode, a high abundance of molecular ions can be obtained, which is yet another possibility for compound identification. Furthermore, by using selected ion monitoring (SIM), molecular ions or molecular fragment ions can be monitored in order to provide a highly sensitive compound-selective detection. Nevertheless, due to limited mass resolution, information on elemental composition is not easily obtained with bench-top MS instrumentation. Therefore, screening of trace levels of heteroatom-containing compounds in complex samples has been more successfully performed by utilizing element-selective detection, e.g., as provided by the atomic emission detector (AED). The only GC-AED instrument on the market today is the Hewlett-Packard G2350A, which has an impressive list of merits in terms of detection sensitivity and element-to-carbon selectivities for a great number of elements.1Plasma mass spectrometry has been coupled with gas chromatography2,3 and has provided a detection performance similar to the AED, but it is usually associated with much lower detection limits. In such instrumentation, several plasmas have been explored as ion sources, including inductively coupled plasmas (ICP),4–12 microwave induced plasmas (MIP)13–18 and radiofrequency glow discharges (rf-GD).19,20 Typical elements of these are the pressure-reducing sampler and skimmer interfaces, which are used to separate the atmospheric or low pressure plasmas from the high vacuum area of the MS. Recently, a microplasma (MP) ion source was introduced by our group.21–25 In contrast to the other plasma ion sources, the MP ion source could be located directly inside the high vacuum area of the MS, due to its very small plasma gas flow. The ion source was a capacitively coupled 350 kHz radiofrequency helium plasma, which was contained inside a 0.3 mm id fused silica capillary tube. One external ring electrode was utilized for applying the rf voltage, which generated a plasma inside the capillary between two grounded external electrodes on both sides of the rf electrode. The plasma could be sustained with only the GC carrier gas (2–3 mL min−1 of helium), which was fed into the capillary ion source, either by using the 4 cm end of the GC capillary column as an ion source,22–24 or by connecting the GC column end to a separate fused silica capillary tube.25 Positively charged atomic ions were expelled from the tip of the capillary into the high vacuum chamber and directly transferred to the quadrupole mass analyzer by electrostatic lenses. Hence, it was concluded that the setup for GC-MP-MS was compatible with bench-top GC-MS instrumentation of limited pumping capacity. Furthermore, the low plasma gas consumption was advantageous, since it provided much lower operational costs than are possible with conventional ICP-MS instrumentation.
For tin-selective detection with GC-MP-MS, relatively large amounts of hydrogen (0.15–1.5 mL min−1) were added to the plasma in order to avoid carbon deposition and tin-to-silica interactions with the surface of the ion source.25 For halogen-selective detection, on the other hand, much smaller amounts of reagent gas were needed (oxygen or a mixture of oxygen and hydrogen at the 1–70 µL min−1 level).23,24 In general, the detection limits for the halogens were at the low picogram level, and the halogen-to-hydrocarbon selectivities were above 104 for chlorine, bromine, and iodine. Unfortunately, fluorine exhibited a poor selectivity to hydrocarbon compounds (8 × 102) in the positive ion detection mode.24 This was caused by interfering H3O+ at m/z 19, which probably originated from the hydrogen and oxygen that were used as reagent gases. In addition, hydrogen was released from eluting hydrocarbon compounds. Although an increase of the power level was applied to reduce this interference, it was never completely eliminated. Therefore, the present study focuses on GC-MP-MS with negative ion detection of the halogens as a solution to overcome the H3O+ interference problem, since the mass spectral background in the negative ion mode was believed to be free of any interfering species at m/z 19. Furthermore, the general halogen-selective detection performance was explored, mainly for the purpose of obtaining lower detection limits.
Experimental
Gas chromatography
Gas chromatography was performed with a GC17A (Shimadzu, Kyoto, Japan), which was equipped with electronic pressure control. The separating fused silica capillary column was a 30 m × 0.32 mm id CP-Sil 8 CB-MS (Chrompack, Middelburg, The Netherlands), which was coated with a 0.25 µm film of 5% phenyl- and 95% dimethylpolysiloxane. Samples of 1.0 µL were injected either splitless or in the split mode (split ratio 1∶9) at an injector temperature of 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. The carrier gas was 99.9999% helium (Aga, Oslo, Norway) at a flow rate of 2 mL min−1. A 1.5 m × 0.25 mm id CP-Sil 8 CB-MS fused silica capillary column (Chrompack), coated with a 0.25 µm film of 5% phenyl- and 95% dimethylpolysiloxane, was used as a transfer tube from the column outlet to the MS. This capillary was contained in a laboratory-built heated and isolated transfer line, which was made of 0.7 mm id and 1/16″ od steel tubing and heated to 300°C by a Model E-03122-61 flexible heating cord (Cole Parmer, Niles, IL, USA), in combination with the auxiliary temperature controller of the gas chromatograph. Near the MS entrance, the capillary was connected to a stainless steel 1/16″ tee (ZT1, VICI, Houston, TX, USA) for reagent gas introduction (Fig. 1). The reagent gases were introduced through a 10 cm × 50 µm id fused silica restrictor (Polymicro Technologies, Phoenix, AZ, USA) from another 1/16″ tee, which allowed two gases to be mixed from two 2.0 m × 30 µm id fused silica restrictors (99.95% oxygen and 99.995% hydrogen, Aga). Each of the reagent gas flows (0.5–30 µL min−1 of oxygen and 0.5–50 µL min−1 of hydrogen) was controlled by the inlet pressure (0–3 bar) prior to the restrictors and calibrated against off-line measurements at different pressures.
°C. The carrier gas was 99.9999% helium (Aga, Oslo, Norway) at a flow rate of 2 mL min−1. A 1.5 m × 0.25 mm id CP-Sil 8 CB-MS fused silica capillary column (Chrompack), coated with a 0.25 µm film of 5% phenyl- and 95% dimethylpolysiloxane, was used as a transfer tube from the column outlet to the MS. This capillary was contained in a laboratory-built heated and isolated transfer line, which was made of 0.7 mm id and 1/16″ od steel tubing and heated to 300°C by a Model E-03122-61 flexible heating cord (Cole Parmer, Niles, IL, USA), in combination with the auxiliary temperature controller of the gas chromatograph. Near the MS entrance, the capillary was connected to a stainless steel 1/16″ tee (ZT1, VICI, Houston, TX, USA) for reagent gas introduction (Fig. 1). The reagent gases were introduced through a 10 cm × 50 µm id fused silica restrictor (Polymicro Technologies, Phoenix, AZ, USA) from another 1/16″ tee, which allowed two gases to be mixed from two 2.0 m × 30 µm id fused silica restrictors (99.95% oxygen and 99.995% hydrogen, Aga). Each of the reagent gas flows (0.5–30 µL min−1 of oxygen and 0.5–50 µL min−1 of hydrogen) was controlled by the inlet pressure (0–3 bar) prior to the restrictors and calibrated against off-line measurements at different pressures. | ||
| Fig. 1 Microplasma ion source. | ||
Mass spectrometry
The mass spectrometer was a modified Model 201 Dedicated Thermospray LC-MS (Vestec, Houston, TX, USA), from which the thermospray probe, tip heater, sampling cone, and discharge electrodes were removed. The MS consisted of a quadrupole mass analyzer (Hewlett-Packard, Wilmington, DE, USA), a Model 342 Channeltron electron multiplier (Detector Technology, Sturbridge, MA, USA), control electronics from the Hewlett-Packard Model 5970 MSD and a Hewlett-Packard 59970C Pascal Series ChemStation with Rev. 3.1.1 MS-MSD software. The mass range was 3–800 u with both positive and negative ion detection included. It was possible to monitor ions as low as m/z 1 by increasing the mass offset parameter. In this work, mainly negative ion detection was used in the selected ion monitoring (SIM) mode. The data files were copied to floppy disks and transferred to a PC by version A.00.03 of the MS-DOS program Lifutil.exe (Hewlett-Packard), and subsequently viewed by version C.03.00 of the MS-Windows program G1034C (Hewlett-Packard).In order to obtain optimum detection performance of iodine, it was necessary to tune and calibrate the MS at m/z values up to 127. Therefore, xenon was introduced in trace amounts through the septum of the injector by a fused silica linear restrictor (10 µm id). The familiar isotope pattern of xenon was observed in the mass spectrum at m/z 124–136, and applied for tuning of the MS electrostatic lenses and for adjustment of the mass scale.
Microplasma ion source
The microplasma ion source (Fig. 1) was mounted on a transparent acrylic disc of 50 mm diameter and 10 mm thickness, and was introduced to the MS high vacuum region as a probe. Similar to the setup which was used earlier,25 two 4 mm holes were drilled in the middle of the disc 8 mm apart, center to center, in which two Teflon plugs were fitted. A 10 cm × 0.5 mm id (1/16″ od) steel tube was placed in one of them. This tubing was electrically grounded and contained the fused silica capillary column from the GC. The mixture of reagent gases was introduced at the 1/16″ tee, which was fixed to the GC column by an FS1.4-5 Valcon ferrule (VICI) and to the steel tubing by an SF-100-VG1 ferrule (Alltech, Deerfield, IL, USA). Contrary to the previous setup, the reagent gas flow was led through the space between the GC column and the steel tubing and introduced to the carrier gas flow at the GC column end. The other Teflon plug contained a 10 cm × 1.6 mm od steel rod. This was connected to the rf electrode and carried the rf voltage from the generator, which was either a HPG-2 (ENI, Rochester, NY, USA) that provided 4 impedance levels, 0–150 W of power and a frequency range of 125–375 kHz, or an AG0201HV-U00 (T&C Power Conversion, Rochester, NY, USA) that provided 4 impedance levels, 0–20 W of power and a frequency range of 100–500 kHz. The plasma was generated between the rf ring electrode and two grounded ring electrodes. The electrodes were made of steel wire, which were twisted around a 4 cm long, 1.65 mm od, 0.35 mm id fused silica tube (Novo-Quartz, Langewiesen, Germany). This tube was the microplasma ion source in which the plasma was sustained, and was connected to the steel tubing by a 1 cm long, 1/8″ od, 1/16″ id Teflon tube. The fused silica tube had a short converging section at the tip with an internal diameter of about 100 µm, which was believed to decrease the divergence of the plasma spray.26 The focusing of the ions was performed by the repeller and electrostatic lenses towards the quadrupole ion trajectory at a 90° angle to the MP ion source probe. The narrowing in the tip of the capillary tube was produced by mounting the tube in an electric drill, followed by heating of the tip in a hydrogen–oxygen flame, in order to achieve symmetrical melting.Chemicals
High purity 1,2-dichlorobenzene, iodobenzene, hexachloroethane, 1-bromooctane, 1,5-dibromopentane and 1-bromodecane were obtained from Fluka AG (Buchs, Switzerland). High purity iodoheptane was obtained from Koch-Light Laboratories (Colnbrook, Buckinghamshire, UK). High purity 1,4-dibromobenzene was obtained from the Eastman Kodak Company (Rochester, NY, USA). Solutions of appropriate concentrations were prepared by using cyclohexane (Rathburn, Walkerburn, UK) as the solvent.Estimation of selectivity and detection limits
The selectivity of each of the halogens to carbon was defined as the ratio of response per mole of halogen to the response per mole of carbon on the halogen-selective chromatograms.27 Detection limits were defined as the amount of element required to produce a signal two times the noise level divided by the peak width at half height.27Results and discussion
Practical considerations
A slightly modified setup for microplasma mass spectrometry was utilized in the present study. The mixture of reagent gases (oxygen and hydrogen) was introduced into the plasma gas (GC carrier gas) at the MP ion source inlet by means of a tee connection on the outside of the MS. This allowed one whole piece of the GC column to be used as a transfer line all the way to the ion source, and the possibility of oxygen induced damage to the stationary phase of the GC column was eliminated. A separate fused silica capillary tube was used as the MP ion source. The tube was easily replaceable, and could be rinsed by methanol sonication in a few cases when contaminated with small particles. The narrowing at the capillary outlet was considered an important parameter for the successful operation of the GC-MP-MS setup. This was deduced from preliminary experiments with capillaries of different orifice diameters.The fused silica capillary tube was surrounded by two grounded ring electrodes with one rf ring electrode in the middle. One of the grounded electrodes was located exactly at the outlet of the MP ion source, while the other electrode made up the starting point of the first plasma zone. The rf electrode could be moved along the capillary tube in order to alter the length of the two plasma zones.
The capacitively coupled plasma inside the capillary tube was sustained by the rf generator, which was set up with the impedance and frequency levels which gave the least amount of reflected power (HPG-2: 1 W, AG0201HV-U00: 0.2 W). The plasma gas was the GC carrier gas, which was kept at 2 mL min−1 for chromatographic reasons and in order to be adapted to the limited pumping capacity of the MS. Generally, the plasma was turned off before the solvent elution, since much higher power levels were required in order to avoid plasma quenching when solvents were introduced. When the plasma was turned on after the solvent elution, it could be observed through the transparent disc. Such a visual inspection was useful in cases of air leaks to the GC carrier line, when the color of the plasma shifted towards violet. It was vital to avoid air leaks, since a small amount of air would behave like any other reagent gas and have a significant impact on the plasma properties.
Negative ion detection
The interference at m/z 19 (H3O+) was clearly present in the positive ion background mass spectrum of the microplasma [Fig. 2(a)]. In the negative mode, however, the same mass region contained less background ions, and none at m/z 19 [Fig. 2(b)]. In fact, the whole negative background spectrum had ions only at m/z 1, 16, 17 and 32, which were assigned to H−, O−, OH− and O2−, respectively. In light of this, negative ion detection was applied for halogen-selective detection, mainly in order to achieve a higher selectivity of fluorine to hydrocarbon compounds, but also to explore the possibility of increased sensitivity for all of the halogens. An early report on negative ion detection in ICP-MS by Vickers et al. showed that at least fluorine and chlorine could be detected with a higher signal-to-noise level than was achieved in the positive mode.28 | ||
| Fig. 2 Background mass spectra (m/z 11–21) of the microplasma in (a) positive mode and (b) negative mode. | ||
Electron attachment processes have been described by Christophorou et al.,29 and a later treatment by Stoffels et al. discussed the mechanisms of negative ion formation in low pressure discharges.30
Resonant electron attachment:
 | (1) |
Deactivation:
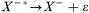 | (2) |
Dissociation:
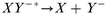 | (3) |
High energy electron collisions:
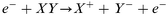 | (4) |
Charge transfer:
 | (5) |
Usually, the first step in negative ion formation is resonant electron attachment to a neutral species (eqn. 1), with the creation of an excited, negatively charged, species. Stable negative ions can then be formed either by collisional or radiative deactivation, in order to lose the excess energy ε (eqn. 2), or by dissociation (eqn. 3). These processes are referred to as non-dissociative and dissociative electron attachment (DA), respectively. An important detail to mention here is that the cross sections for dissociative attachment of halogen molecules decrease in the order I2 > Br2 > Cl2 > F2, and that the same trend is valid for other halogen-containing molecules like CCl4, CF4, etc.29 Negative ionization by dissociative electron attachment should therefore be most important for iodine compounds and least important for fluorine compounds. Also, other mechanisms have been proposed for negative ion formation, e.g., the non-resonant process of high energy electron collisions with molecules or the charge transfer between anions and molecules. Charge transfer is energetically favored if the ionized species has a higher electron affinity than the ionizing species. Proton abstraction should also be considered31 (from HF, HCl, HBr and HI) by exothermic reactions with H−, O−, and OH−. Nevertheless, according to Stoffels et al.,30 dissociative electron attachment is the most important negative ion formation mechanism in low pressure plasmas. Furthermore, the three most important destruction mechanisms are ion–ion recombination (eqn. 6), electron detachment by neutrals (eqn. 7–9) and electron detachment by electrons (eqn. 10). Additionally, wall losses might occur, in which the negative ions diffuse to the ion source wall, followed by neutralization.
Ion–ion recombination:
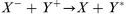 | (6) |
Electron detachment by neutrals:
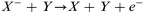 | (7) |
 | (8) |
 | (9) |
Electron detachment by electrons:
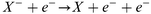 | (10) |
It is important to consider both the formation and the destruction of negative ions in the microplasma, in order to predict the halogen ion abundance. For instance, owing to their high electron affinities, halogens might follow the charge transfer route (eqn. 5), and exhibit ion abundances that are dependent on reagent gas anion densities (H−, O−, etc.). On the other hand, high abundances of positively charged ions (He+, H+, O+, etc.) might lead to increased destruction of the halogen anions. The rate constant (krec) of negative ion breakdown caused by ion–ion recombinations (eqn. 6) can be approximated by:30
 | (11) |
Here, εa is the electron binding energy of the negative ion in eV, T is the temperature in K, µ is the reduced mass of the ions in u and kb is the Boltzmann constant. From this equation, it is interesting to note that the small, positively charged, hydrogen and helium ions will have a greater impact on the anion destruction rate (krec) than the larger positively charged oxygen ions (a factor of 3.0 for H+versus O+ and a factor of 1.6 for He+versus O+). A decrease in the halogen anion abundance should therefore be expected with an increased power level, which produces more He+. Furthermore, the breakdown rate increases with decreasing mass of the halogen anions, and is highest for fluorine. Therefore, if ion–ion recombination is the main breakdown route for the halogen anions, and if similar formation rates are obtained for the different halogens, iodine should exhibit the highest abundance.
Plasma source configuration and power level
In the present study, the microplasma was explored as a source of negatively charged atomic ions. As predicted, the halogen anion abundance at the MP outlet was largely affected by the power level, and found to decrease with an increase of power. In fact, only 1–2 W of power were used, which was lower than the power levels of optimum halogen signal in the positive ion detection mode (3–7 W).23,24 Also, the length of the second plasma zone (Fig. 1, P2) was found to be an important parameter. Plasma lengths (P2) of 7, 5, 3 and 1.5 mm were applied to find the optimum detection performance for fluorine. Each plasma required its own optimization of power: 1.5 mm gave the highest signal-to-noise ratio of fluorine (Fig. 3). As the plasma length was decreased, less power was required. This was explained by an extension of the plasma spray out of the MP ion source, when the rf electrode was moved closer to the ion source outlet, while the generator was set to equal power levels. Therefore, in order to obtain an equally extended plasma spray, the power had to be reduced when reducing P2. The lower power level seemed advantageous for fluorine-selective detection. This could be accounted for by more low-energy electrons produced in the cooler plasma, which would facilitate the electron attachment process (eqn. 1 and 2 or eqn. 1 and 3). Additionally, a reduced decomposition rate was probable, as fewer positively charged species were produced to induce ion–ion collisions (eqn. 6). Finally, the possibility of less radial fluorine diffusion to the ion source walls should be mentioned as an explanation, although fewer fluorine-to-silica interactions (to give peak tailing) were encountered with the low power levels utilized in this work than with the higher power levels utilized in the positive mode.23,24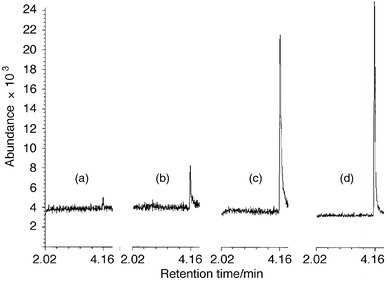 | ||
| Fig. 3 Fluorine-selective chromatograms of 1-fluoronaphthalene (0.75 ng F introduced) at an oxygen flow of 5 µL min−1 and a hydrogen flow of 2 µl min−1, and with a second plasma (Fig. 1, P2) length of (a) 7 mm, (b) 5 mm, (c) 3 mm and (d) 1.5 mm. Forward power levels: (a) 1.96, (b) 1.46, (c) 1.10, and (d) 0.97 W. GC conditions: 1 µL split injection (1∶10) at 110°C. | ||
It was also noticed that an increase in the fluorine signal was obtained with an increased oxygen level. However, at these low power levels, and with a P2 length of 7 or 5 mm, the oxygen tolerance of the plasma was low in terms of plasma quenching during the elution of the compounds. With 1.5 mm, on the other hand, up to 50 µL min−1 of oxygen could be applied without extinguishing the plasma. Therefore, further experiments were carried out with a P2 length of 1.5 mm.
Reagent gases and power level
The introduction of oxygen as reagent gas was vital for the detection of the halogens. At low oxygen levels, no fluorine signal was observed. Fig. 4 shows the combined effect of the oxygen level and the forward power level on the fluorine signal, which indicates an increase of the signal with increased oxygen flow rates. Indeed, as much as 30 µL min−1 of oxygen could be applied to obtain a high signal. This corresponded well with the charge transfer mechanism (eqn. 5) for halogen anion formation by reaction with O−. However, the possibility of obtaining low-energy electrons by oxygen cooling of the plasma should not be ruled out either, as this would facilitate the electron attachment process (eqn. 1 and 2 or eqn. 1 and 3). | ||
| Fig. 4 Effect of oxygen level and forward power on fluorine signal (0.75 ng F introduced). No hydrogen applied. | ||
By introducing trace levels of hydrogen in addition to the oxygen, the fluorine signal was observed to increase (Fig. 5), probably as a result of introduction of H− ions, which might donate electrons in a charge transfer process (eqn. 5). However, at further increased hydrogen levels the fluorine signal decreased rapidly. The latter was explained by a higher density of small positively charged ions (H+), which probably increased the breakdown rate (eqn. 11) of the ion–ion recombination reactions (eqn. 6) with the halogen anions. The highest fluorine response was obtained with a hydrogen flow rate in the 1.5–2.0 µL min−1 range combined with an oxygen flow rate in the 20–25 µL min−1 range.
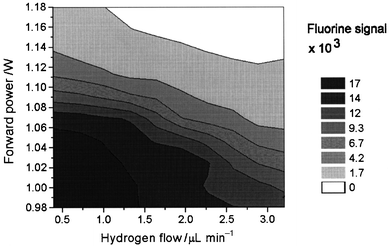 | ||
| Fig. 5 Effect of hydrogen level and forward power on fluorine signal (0.75 ng F introduced): 23 µL min−1 of oxygen applied. | ||
Detection limits and selectivity
Chlorine, bromine and iodine could also be detected with the fluorine-optimized GC-MP-MS setup (Fig. 6). These halogens exhibited more than 10 times improved detection limits in the negative mode (Table 1) than was possible in the positive mode.23,24 With the observation of increased response per mole of halogen with increased halogen size (Table 1), it was deduced that electron attachment (eqn. 1 and 2 or 1 and 3) was the most important mechanism for the larger halogens. The differences in the responses also supported the prediction of the breakdown rates being higher for fluorine anions than for the larger halogen anions. Furthermore, owing to its electronegativity, fluorine possibly followed an ion collision charge transfer process (eqn. 5), which is less kinetically favored than electron attachment.31 This could explain the much lower response for fluorine compared with iodine. | ||
| Fig. 6 Chlorine-, bromine-, and iodine-selective chromatograms (72 pg of halogen per compound) of 1,2-dichlorobenzene, iodobenzene, hexachloroethane, iodoheptane, 1-bromooctane, 1,4-dibromobenzene, 1,5-dibromopentane and 1-bromodecane (in eluting order). GC conditions: 1 µL splitless injection at 50°C, held for 5 min, then temperature programmed to 200°C at 10°C min−1, while pressure programmed at 4 kPa min−1 to keep a constant mass flow of carrier gas. Reagent gas flows: 21 µL min−1 of oxygen and 1.6 µL min−1 of hydrogen. | ||
| Compound | Element | m/z | Element limit of detection/pg s−1 | Isotope limit of detection/pg s−1 | Response per mole of halogen isotope relative to response per mole of fluorine |
|---|---|---|---|---|---|
| 1-Fluoronaphthalene | F | 19 | 12 | 12 | 1 |
| Hexachloroethane | Cl | 35 | 0.35 | 0.27 | 77 |
| 1,4-Dibromobenzene | Br | 79 | 0.50 | 0.25 | 210 |
| Iodoheptane | I | 127 | 0.13 | 0.13 | 581 |
Halogen selectivities to carbon (Table 2) showed an increase with atomic mass number. This was accounted for by both increased response and decreased background level with increasing mass of the halogens. As predicted from the examination of the background mass spectra, the selectivity of fluorine to carbon was much higher in the negative mode than in the positive mode24 (by a factor of 4).
| Element | Selectivity |
|---|---|
| F | 3 × 103 |
| Cl | 2 × 104 |
| Br | 2 × 105 |
| I | 8 × 105 |
Conclusion
Gas chromatography coupled with microplasma mass spectrometry has been applied for the detection of negatively charged halogen atoms. The possibility of using negative ion detection to completely avoid the background interference problem at m/z 19 for fluorine-selective detection was demonstrated. Thus, it was possible to detect fluorine with both high sensitivity and selectivity to hydrocarbon compounds. Additionally, the simultaneous detection of the halogens revealed a high sensitivity also for chlorine, bromine and iodine.Acknowledgements
Professor Einar Uggerud at University of Oslo, Norway, and Dr. Eva Stoffels at Eindhoven University of Technology, The Netherlands, are acknowledged for helpful discussions.References
- B. D. QuimbyP. A. LarsonP. C. Dryden, Application Note 228-363, Publication Number 5965-4720E, Hewlett-Packard, Wilmington, DE, USA, 1996..
- N. P. Vela, L. K. Olson and J. A. Caruso, Anal. Chem., 1993, 65, 585.
- R. Lobinski and F. C. Adams, Spectrochim. Acta, Part B, 1997, 52, 1865 CrossRef.
- N. S. Chong and R. S. Houk, Appl. Spectrosc., 1987, 41, 66 Search PubMed.
- A. Kim, S. Hill, L. Ebdon and S. Rowland, J. High Resolut. Chromatogr., 1992, 15, 665 CAS.
- A. W. Kim, M. E. Foulkes, L. Ebdon, S. J. Hill, R. L. Patience, A. G. Barwise and S. J. Rowland, J. Anal. At. Spectrom., 1992, 7, 1147 RSC.
- E. H. Evans and J. A. Caruso, J. Anal. At. Spectrom., 1993, 8, 427 RSC.
- T. M. Castillano, J. J. Giglio, E. H. Evans and J. A. Caruso, J. Anal. At. Spectrom., 1994, 9, 1335 RSC.
- E. H. Evans, W. Pretorius, L. Ebdon and S. Rowland, Anal. Chem., 1994, 66, 3400 CrossRef CAS.
- A. Prange and E. Jantzen, J. Anal. At. Spectrom., 1995, 10, 105 RSC.
- G. O'Connor, L. Ebdon, E. H. Evans, H. Ding, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 1151 RSC.
- T. De Smaele, L. Moens, R. Dams and P. Sandra, Fresenius' J. Anal. Chem., 1996, 355, 778 CAS.
- J. T. Creed, A. H. Mohamad, T. M. Davidson, G. Ataman and J. A. Caruso, J. Anal. At. Spectrom., 1988, 3, 923 RSC.
- A. H. Mohamad, J. T. Creed, T. M. Davidson and J. A. Caruso, Appl. Spectrosc., 1989, 43, 1127 Search PubMed.
- H. Suyani, J. Creed, J. A. Caruso and R. D. Satzger, J. Anal. At. Spectrom., 1989, 4, 777 RSC.
- J. T. Creed, T. M. Davidson, W. L. Shen and J. A. Caruso, J. Anal. At. Spectrom., 1990, 5, 109 RSC.
- W. C. Story, L. K. Olson, W. L. Shen, J. T. Creed and J. A. Caruso, J. Anal. At. Spectrom., 1990, 5, 467 RSC.
- W. C. Story and J. A. Caruso, J. Anal. At. Spectrom., 1993, 8, 571 RSC.
- L. K. Olson, M. Belkin and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 491 RSC.
- M. A. Belkin, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1997, 12, 1255 RSC.
- C. BredeS. Pedersen-BjergaardE. LundanesT. Greibrokk, Patent pending, International Patent Application serial number PCT/NO98/00048. .
- C. Brede, S. Pedersen-Bjergaard, E. Lundanes and T. Greibrokk, Anal. Chem., 1998, 70, 513 CrossRef CAS.
- C. Brede, E. Lundanes, T. Greibrokk and S. Pedersen-Bjergaard, J. High Resolut. Chromatogr., 1998, 21, 282 CrossRef CAS.
- C. Brede, E. Lundanes, T. Greibrokk and S. Pedersen-Bjergaard, J. High Resolut. Chromatogr., 1998, 21, 633 CrossRef CAS.
- C. Brede, S. Pedersen-Bjergaard, E. Lundanes and T. Greibrokk, J. Chromatogr. A, 1999, 849, 553 CrossRef CAS.
- H. R. Murphy and D. R. Miller, J. Phys. Chem., 1984, 88, 4474 CrossRef CAS.
- B. D. Quimby and J. J. Sullivan, Anal. Chem., 1990, 62, 1027 CrossRef CAS.
- G. H. Vickers, D. A. Wilson and G. M. Hieftje, Anal. Chem., 1988, 60, 1808 CrossRef CAS.
- L. G. ChristophorouD. L. McCorckleA. A. Christodoulides, in Electron–Molecule Interactions and Their Applications, ed. L. G. Christophorou, Academic Press, San Diego, CA, USA, 1984, vol. 1. Search PubMed.
- E. Stoffels, W. W. Stoffels, D. Vender, M. Haverlag, G. M. W. Kroesen and F. J. De Hoog, Contrib. Plasma Phys., 1995, 35, 331 CAS.
- A. G. Harrison, in Chemical Ionization Mass Spectrometry, CRC Press, Boca Raton, FL, USA, 2nd edn., 1992, ch. 2, pp. 24–40. .
| This journal is © The Royal Society of Chemistry 2000 |