The changing nature of the 206Pb/207Pb isotopic ratio of lead in rainwater, atmospheric particulates, pine needles and leaded petrol in Scotland, 1982–1998
John G. Farmer*a, Lorna J. Eadesa, Margaret C. Grahama and Jeffrey R. Baconb
aDepartment of Chemistry, The University of Edinburgh, West Mains Road, Edinburgh, UK EH9 3JJ
bSoil Science Group, Macaulay Land Use Research Institute, Craigiebuckler, Aberdeen, UK AB15 8QH
First published on UnassignedUnassigned1st February 2000
Abstract
The inductively coupled plasma-mass spectrometry (ICP-MS)-determined 206Pb/207Pb ratio of 145 samples of rainwater collected at 25 locations around Scotland during December 1997 and January 1998 and at three long-term monitoring stations in the northeast, central belt and southeast of the country from November 1997 to December 1998 averaged 1.144 ± 0.017 (1 s). This represents a significant increase from the mean value of 1.120 ± 0.016 recorded for the long-term sites in 1989–1991, only partly attributable to a concomitant increase in the 206Pb/207Pb ratio of leaded petrol from 1.075 ± 0.013 to 1.088 ± 0.007. The rainwater 206Pb/207Pb data for the late 1990s also contrast markedly with the lower 206Pb/207Pb ratios found for pine needle and atmospheric particulate samples from Scotland (e.g. Glasgow: 1.085 ± 0.012 in 1985–1986, 1.099 ± 0.007 in 1991–1992), England and Western Europe in this study for the period 1982–1992 when emissions of lead to the atmosphere from petrol-engined vehicles in the UK were ∼2–9 times higher. The observed change in the lead isotopic signature of rainwater predominantly reflects the impact of measures, such as the introduction and growing uptake of unleaded petrol, to reduce car exhaust emissions of lead to the atmosphere in the UK. Based on the rainwater data, source apportionment calculations suggest a general decline in the contribution of leaded petrol to atmospheric lead in Scotland from 53–61% in 1989–1991 to 32–45% in 1997–1998, with a corresponding decline in the urban environment from 84–86% to 48–58%.
Introduction
The aim of this study was to determine the stable lead isotopic signature (206Pb/207Pb) of rainwater in Scotland in the late 1990s and to compare the findings with corresponding data for rainwater and atmospheric particulates collected since the 1980s and for different sources of environmental lead, with a view to evaluating the impact of measures to reduce emissions of lead to the atmosphere, such as the introduction and increasing use of unleaded petrol.There are four stable isotopes of primordial lead, namely 204Pb, 206Pb, 207Pb and 208Pb. The last three isotopes, however, are also radiogenic, formed by radioactive decay in the 238U (half-life 4.5 × 109 years), 235U (half-life 0.7 × 109 years) and 232Th (half-life 14.1 × 109 years) decay series, respectively. The abundance of lead isotopes in minerals and ores is determined by the original uranium to thorium ratio, time of formation and lead concentration of the material. While the average present-day crustal abundances of these isotopes yield a mean 206Pb/207Pb ratio of 1.20 ± 0.015, there is a much wider range found for lead ore deposits around the world, e.g. 1.04 for Broken Hill, Australia, 1.17 for Leadhills, Scotland, and 1.39 for Mississippi Valley, MO, USA.1,2
The lead ore deposits from Australia have been used, along with Canadian lead (206Pb/207Pb = 1.16), by Associated Octel to produce alkyllead anti-knock additives (206Pb/207Pb ∼ 1.07) for petrol consumed in the UK.3 The significant difference between the 206Pb/207Pb ratio of the lead emitted by car exhausts and those of other sources of anthropogenic lead, e.g. coal burning (206Pb/207Pb ∼ 1.18), has afforded an opportunity, at least in principle, for the quantitative assessment of the relative contributions from specific sources to the environmental lead burden.4–6 Thus, 206Pb/207Pb ratio variations have been recorded and used in numerous parts of the world, where different characteristic isotopic signatures may prevail, in such studies on a wide range of materials, including atmospheric aerosols,4,7–14 lake sediments,15–17 peat bogs,17–20 soils,21–23 grass,24 tree rings,25,26 moss27,28 and ice cores.29,30 They have also been used to trace air mass movement31–33 and oceanic transport phenomena34 as well as the environmental fate and behaviour of anthropogenic lead.35–38
In the UK, measures introduced in 1986 to reduce the maximum permitted concentration of lead in petrol from 0.40 to 0.15 g l−1 and to promote the use of unleaded petrol have resulted in a reduction of emissions of lead from petrol-engined vehicles to the atmosphere from an estimated 6.5 × 103 tonnes in 1985 to 0.8 × 103 tonnes in 1997.39 Resultant changes in source apportionment of atmospheric lead with time and the use of the 206Pb/207Pb ratio as a tracer are dependent upon the monitoring of stable lead isotopic ratios in the atmosphere and in contributory sources such as leaded petrol. This paper presents isotopic data for rainwater in Scotland in 1997–1998, directly determined by the technique of inductively coupled plasma-mass spectrometry (ICP-MS),40 and interprets them in the context of additional long-term data for leaded petrol from 1989–1998 and for rainwater, atmospheric particulates and pine needles collected at various locations in Scotland and elsewhere during the period 1982–1992.
Experimental
Sample collection and preparation
Petrol samples (100 ml) (n = 33) were collected directly at brand-name pumps in Edinburgh on dates between February 1989 and November 1998 as shown in Table 1 and the lead was extracted using the iodine monochloride/solvent extraction method of Price,41 prior to final solution preparation in 2% v/v (0.32 M) nitric acid for analysis.| Brand of petrol | Date of collection | 206Pb/207Pb (±1 s) |
|---|---|---|
| aThe Oct. 1994 petrol sample was collected in Ullapool, northwest Scotland. | ||
| Feb. 1989 | 1.056 ± 0.001 | |
| Dec. 1989 | 1.066 ± 0.007 | |
| Mean | (1989) | 1.061 ± 0.0071 |
| BP | Dec. 1990 | 1.069 ± 0.002 |
| Jet | Dec. 1990 | 1.074 ± 0.003 |
| Shell | Dec. 1990 | 1.062 ± 0.004 |
| Texaco | Dec. 1990 | 1.083 ± 0.001 |
| Mean | (1990) | 1.072 ± 0.0088 |
| Savacentre | May 1991 | 1.093 ± 0.003 |
| Savacentre | Dec. 1991 | 1.088 ± 0.002 |
| Texaco | Dec. 1991 | 1.084 ± 0.002 |
| Mean | (1991) | 1.088 ± 0.0045 |
| Savacentre | Nov. 1993 | 1.070 ± 0.003 |
| Gulf | Dec. 1993 | 1.067 ± 0.003 |
| Gulf | Jan. 1994 | 1.067 ± 0.005 |
| Savacentre | Jan. 1994 | 1.067 ± 0.004 |
| Texaco | Jan. 1994 | 1.075 ± 0.002 |
| Mean | (1993–1994) | 1.069 ± 0.0035 |
| BP | Oct. 1994a | 1.063 ± 0.003 |
| Gulf | Nov. 1994 | 1.062 ± 0.002 |
| Jet | Nov. 1994 | 1.068 ± 0.003 |
| Texaco | Nov. 1994 | 1.065 ± 0.003 |
| Mean | (1994) | 1.065 ± 0.0026 |
| BP | Feb. 1996 | 1.076 ± 0.001 |
| Jet | Feb. 1996 | 1.092 ± 0.003 |
| Shell | Feb. 1996 | 1.080 ± 0.002 |
| Mean | (1996) | 1.083 ± 0.0083 |
| BP | Mar. 1997 | 1.074 ± 0.005 |
| Esso | Mar. 1997 | 1.076 ± 0.004 |
| Savacentre | Mar. 1997 | 1.073 ± 0.001 |
| Shell | Mar. 1997 | 1.072 ± 0.005 |
| Mean | (Early 1997) | 1.074 ± 0.0017 |
| Safeway | Nov. 1997 | 1.083 ± 0.0017 |
| Savacentre | Nov. 1997 | 1.097 ± 0.0011 |
| Shell | Nov. 1997 | 1.098 ± 0.0014 |
| Texaco | Nov. 1997 | 1.075 ± 0.0013 |
| Mean | (Late 1997) | 1.088 ± 0.0111 |
| BP | Nov. 1998 | 1.085 ± 0.0012 |
| Jet | Nov. 1998 | 1.086 ± 0.0014 |
| Savacentre | Nov. 1998 | 1.088 ± 0.0011 |
| Shell | Nov. 1998 | 1.089 ± 0.0008 |
| Mean | (1998) | 1.087 ± 0.0018 |
| Mean | (n = 33) | 1.076 ± 0.011 |
Rainwater samples were collected by one of three methods. For the samples (n = 47, Table 2) collected during December 1997 and January 1998 from 25 sites around Scotland (Fig. 1), 1 l polypropylene bottles (Merck, Poole, Dorset, UK), each with a 14 cm diameter polypropylene filter funnel taped to its rim, were placed at least 3 m above the ground, usually in a domestic garden away from obstruction and possible contamination. For the samples (n = 98, Table 3) collected on a regular basis between November 1997 and December 1998 at the three long-term monitoring sites specifically located away from roads and industrial activity at Glensaugh (weekly), Hartwood (monthly) and Sourhope (weekly) (Fig. 1), two methods were used.35 The open gauge method (n = 68) collects rainfall (or anything that drops vertically) using shielded rain gauges designed to remove any effect of turbulence around the gauge and installed in triplicate 3 m above ground level. The filter gauge method (n = 30), which collects not only rainfall but also air particulates and mist that could come in sideways, uses specially adapted gauges installed in triplicate 3 m above ground level and alongside the open gauges. In theory, the open gauge samples should therefore yield the isotopic character of lead being deposited in rainwater, both dissolved and washed out particulates, whereas the filter gauge samples might indicate if there are other components preferentially intercepted by vegetation. The open filter funnel method should be intermediate between these approaches to rainfall collection. After filtering (Whatman 542, 2.7 µm, 18.5 cm diameter Maidstone, UK), sample aliquots were usually concentrated to 10% of their original volume by evaporation on a hotplate at 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C and acidified to 2% v/v nitric acid prior to analysis. It should be noted that most of the rainwater samples (i.e. from Glensaugh, Hartwood and Sourhope) were initially collected not merely for lead but for anions and basic water analysis. Thus they were not treated immediately after collection in any way other than by filtration. Although it is therefore possible that the lead concentrations of these samples may have been affected, e.g. by adsorption to sample container walls, the lead isotopic ratios, the focus of the work presented here, would not have been affected.
°C and acidified to 2% v/v nitric acid prior to analysis. It should be noted that most of the rainwater samples (i.e. from Glensaugh, Hartwood and Sourhope) were initially collected not merely for lead but for anions and basic water analysis. Thus they were not treated immediately after collection in any way other than by filtration. Although it is therefore possible that the lead concentrations of these samples may have been affected, e.g. by adsorption to sample container walls, the lead isotopic ratios, the focus of the work presented here, would not have been affected.
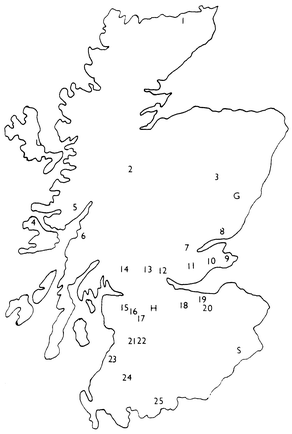 | ||
| Fig. 1 Map of Scotland showing the 25 rainwater sampling locations during December 1997 and January 1998 (see Table 2 for key) and the three long-term rainwater collection sites at Glensaugh (G), Hartwood (H) and Sourhope (S). | ||
| December 1997 | January 1998 | |
|---|---|---|
| Location | 206Pb/207Pb (±1 s) | 206Pb/207Pb (±1 s) |
| 1. Reay | 1.160 ± 0.0098 | 1.154 ± 0.0058 |
| 2. Fort Augustus | 1.147 ± 0.0095 | 1.133 ± 0.0074 |
| 3. Tarland | 1.160 ± 0.0035 | 1.144 ± 0.0079 |
| 4. Tobermory | 1.150 ± 0.0090 | 1.154 ± 0.0074 |
| 5. Kilmalieu | 1.152 ± 0.0094 | 1.156 ± 0.0039 |
| 6. Oban | 1.144 ± 0.0049 | 1.149 ± 0.0027 |
| 7. Bridge of Earn | 1.123 ± 0.0023 | |
| 8. Dundee | 1.161 ± 0.0012 | 1.150 ± 0.0092 |
| 9. St. Andrews | 1.151 ± 0.0019 | 1.144 ± 0.0042 |
| 10. Ceres | 1.147 ± 0.0084 | |
| 11. Kinross | 1.128 ± 0.0010 | 1.131 ± 0.0008 |
| 12. Menstrie | 1.131 ± 0.0037 | 1.133 ± 0.0055 |
| 13. Stirling | 1.147 ± 0.0042 | 1.218 ± 0.0072 |
| 14. Buchlyvie | 1.137 ± 0.0032 | 1.112 ± 0.0028 |
| 15. Paisley | 1.147 ± 0.0027 | |
| 16. Giffnock | 1.139 ± 0.0024 | 1.133 ± 0.0037 |
| 17. East Kilbride | 1.141 ± 0.0071 | 1.117 ± 0.0057 |
| 18. Livingston | 1.121 ± 0.0037 | 1.127 ± 0.0081 |
| 19. Edinburgh A | 1.179 ± 0.0031 | 1.186 ± 0.0053 |
| 20. Edinburgh B | 1.155 ± 0.0004 | 1.207 ± 0.0005 |
| 21. Kilmarnock A | 1.143 ± 0.0026 | 1.125 ± 0.0032 |
| 22. Kilmarnock B | 1.139 ± 0.0049 | 1.132 ± 0.0045 |
| 23. Ayr | 1.152 ± 0.0023 | 1.136 ± 0.0059 |
| 24. Loch Bradan | 1.122 ± 0.0017 | 1.119 ± 0.0012 |
| 25. Dumfries | 1.131 ± 0.0030 | 1.123 ± 0.0057 |
| Mean | 1.145 ± 0.014 | 1.144 ± 0.026 |
| Glensaugh | Hartwood | Sourhope | ||||
|---|---|---|---|---|---|---|
| Open gauge | Filter gauge | Open gauge | Filter gauge | Open gauge | Filter gauge | |
| Date of collection | 206Pb/207Pb (±1 s) | 206Pb/207Pb (±1 s) | 206Pb/207Pb (±1 s) | 206Pb/207Pb (±1 s) | 206Pb/207Pb (±1 s) | 206Pb/207Pb (±1 s) |
| 19 Nov. 1997 | 1.140 ± 0.0039 | 1.144 ± 0.0040 | ||||
| 26 Nov. 1997 | 1.168 ± 0.0051 | 1.144 ± 0.0019 | 1.138 ± 0.0036 | 1.145 ± 0.0039 | ||
| 3 Dec. 1997 | 1.154 ± 0.0061 | 1.140 ± 0.0021 | ||||
| 17 Dec. 1997 | 1.163 ± 0.0047 | 1.155 ± 0.0028 | 1.171 ± 0.0041 | 1.149 ± 0.0035 | 1.145 ± 0.0032 | 1.153 ± 0.0133 |
| 14 Jan. 1998 | 1.155 ± 0.0051 | 1.158 ± 0.0016 | ||||
| 21 Jan. 1998 | 1.180 ± 0.0060 | 1.148 ± 0.0043 | 1.148 ± 0.0070 | 1.155 ± 0.0069 | ||
| 28 Jan. 1998 | 1.137 ± 0.0054 | |||||
| 11 Feb. 1998 | 1.161 ± 0.0029 | |||||
| 18 Feb. 1998 | 1.181 ± 0.0118 | 1.118 ± 0.0109 | 1.152 ± 0.0083 | 1.152 ± 0.0074 | ||
| 25 Feb. 1998 | 1.140 ± 0.0053 | |||||
| 11 Mar. 1998 | 1.138 ± 0.0076 | |||||
| 18 Mar. 1998 | 1.207 ± 0.0087 | 1.134 ± 0.0056 | 1.129 ± 0.0035 | |||
| 25 Mar. 1998 | 1.135 ± 0.0054 | |||||
| 1 Apr. 1998 | 1.144 ± 0.0015 | 1.141 ± 0.0041 | ||||
| 8 Apr. 1998 | 1.132 ± 0.0010 | 1.139 ± 0.0033 | 1.135 ± 0.0029 | |||
| 15 Apr. 1998 | 1.141 ± 0.0043 | |||||
| 22 Apr. 1998 | 1.139 ± 0.0017 | 1.139 ± 0.0014 | 1.190 ± 0.0050 | 1.144 ± 0.0018 | 1.142 ± 0.0042 | 1.146 ± 0.0051 |
| 29 Apr. 1998 | 1.140 ± 0.0018 | 1.131 ± 0.0013 | ||||
| 6 May 1998 | 1.140 ± 0.0039 | |||||
| 13 May 1998 | 1.178 ± 0.0015 | 1.143 ± 0.0022 | 1.144 ± 0.0072 | 1.150 ± 0.0030 | ||
| 20 May 1998 | 1.146 ± 0.0098 | |||||
| 27 May 1998 | 1.140 ± 0.0022 | 1.134 ± 0.0045 | ||||
| 3 June 1998 | 1.144 ± 0.0039 | 1.140 ± 0.0029 | 1.142 ± 0.0051 | |||
| 10 June 1998 | 1.140 ± 0.0021 | 1.149 ± 0.0025 | 1.144 ± 0.0020 | 1.137 ± 0.0035 | 1.142 ± 0.0030 | |
| 17 June 1998 | 1.138 ± 0.0029 | 1.141 ± 0.0019 | ||||
| 24 June 1998 | 1.140 ± 0.0034 | |||||
| 1 July 1998 | 1.126 ± 0.0019 | 1.136 ± 0.0030 | ||||
| 8 July 1998 | 1.152 ± 0.0025 | |||||
| 15 July 1998 | 1.141 ± 0.0030 | 1.138 ± 0.0046 | ||||
| 22 July 1998 | 1.136 ± 0.0033 | |||||
| 29 July 1998 | 1.127 ± 0.0041 | |||||
| 5 Aug. 1998 | 1.133 ± 0.0020 | 1.112 ± 0.0035 | ||||
| 12 Aug. 1998 | 1.141 ± 0.0074 | |||||
| 19 Aug. 1998 | 1.145 ± 0.0027 | 1.123 ± 0.0024 | ||||
| 2 Sept. 1998 | 1.135 ± 0.0016 | |||||
| 16 Sept. 1998 | 1.144 ± 0.0020 | |||||
| 30 Sept. 1998 | 1.145 ± 0.0017 | |||||
| 7 Oct. 1998 | 1.156 ± 0.0014 | 1.151 ± 0.0013 | ||||
| 14 Oct. 1998 | 1.153 ± 0.0016 | |||||
| 21 Oct. 1998 | 1.113 ± 0.0016 | 1.141 ± 0.0012 | ||||
| 28 Oct. 1998 | 1.138 ± 0.0006 | 1.140 ± 0.0029 | ||||
| 4 Nov. 1998 | 1.135 ± 0.0024 | |||||
| 11 Nov. 1998 | 1.136 ± 0.0006 | 1.142 ± 0.0033 | ||||
| 18 Nov. 1998 | 1.136 ± 0.0011 | 1.137 ± 0.0014 | ||||
| 25 Nov. 1998 | 1.142 ± 0.0018 | 1.137 ± 0.0020 | ||||
| 2 Dec. 1998 | 1.133 ± 0.0011 | |||||
| 9 Dec. 1998 | 1.138 ± 0.0014 | |||||
| 16 Dec. 1998 | 1.141 ± 0.0014 | |||||
| 23 Dec. 1998 | 1.156 ± 0.0008 | |||||
| Mean | 1.140 ± 0.010 | 1.144 ± 0.008 | 1.164 ± 0.024 | 1.141 ± 0.010 | 1.140 ± 0.008 | 1.147 ± 0.008 |
Oven-dried, powdered pine needle samples (n = 14) for 1982–1986 from the locations in Scotland, England, Netherlands and Germany listed in Table 4 were provided by Dr. J. N. Cape, Natural Environment Research Council Institute of Terrestrial Ecology (NERC ITE), Penicuik. These samples were ashed at 450°C, digested in 8 M nitric acid and the resultant extracts diluted in 2% v/v nitric acid for analysis.
| Location and type of sample | Date of collection | 206Pb/207Pb (±1 s) |
|---|---|---|
| Netherlands* | ||
| Kootwijk | 1982 (growth) | 1.123 ± 0.002 |
| 1983 (growth) | 1.122 ± 0.002 | |
| 1985 (growth) | 1.127 ± 0.008 | |
| Rip | 1984 (growth) | 1.129 ± 0.003 |
| Germany* | ||
| Kalbelescheuer, Black Forest | 1985 (growth) | 1.121 ± 0.006 |
| 1985 (growth) | 1.136 ± 0.007 | |
| N. Langebramke, Hartz Mountains | 1986 (growth) | 1.120 ± 0.003 |
| Selb, E. Bavaria | 1986 (growth) | 1.132 ± 0.012 |
| Mean | (1982–1986) | 1.126 ± 0.006 |
| Scotland* | ||
| Glenbranter, N.W. | 1985 (growth) | 1.106 ± 0.004 |
| 1985 (growth) | 1.117 ± 0.008 | |
| Darnaway, N.E. | 1985 (growth) | 1.111 ± 0.005 |
| 1985 (growth) | 1.117 ± 0.009 | |
| England* | ||
| Ashford, S.E. | 1985 (growth) | 1.117 ± 0.007 |
| 1985 (growth) | 1.115 ± 0.009 | |
| Mean | (1985) | 1.114 ± 0.004 |
| Scotland (urban) | ||
| Glasgow | Jan. 1985 | 1.069 ± 0.004 |
| Apr. 1985 | 1.089 ± 0.003 | |
| Jun. 1985 | 1.083 ± 0.003 | |
| Jun. 1986 | 1.097 ± 0.003 | |
| Mean | (1985–1986) | 1.085 ± 0.012 |
| Scotland (rural) | ||
| Banchory, Grampian | Feb./Mar. 1985 | 1.107 ± 0.007 |
| Mar. 1985 | 1.113 ± 0.003 | |
| Crathes, Grampian | Feb./Mar. 1985 | 1.128 ± 0.009 |
| Apr. 1985 | 1.121 ± 0.006 | |
| Brathens, Grampian | May 1985 | 1.116 ± 0.002 |
| June 1985 | 1.111 ± 0.002 | |
| June/July 1985 | 1.120 ± 0.004 | |
| July/Aug. 1985 | 1.122 ± 0.009 | |
| Aug./Sept. 1985 | 1.119 ± 0.002 | |
| Devilla, Fife | May 1985 | 1.124 ± 0.004 |
| Mean | (1985) | 1.118 ± 0.006 |
| England | ||
| Gisburn, Lancashire | Mar. 1985 | 1.111 ± 0.004 |
| Apr./May 1985 | 1.100 ± 0.003 | |
| Sept. 1985 | 1.088 ± 0.003 | |
| Oct. 1985 | 1.102 ± 0.002 | |
| Nov. 1985 | 1.092 ± 0.003 | |
| Mean | (1985) | 1.108 ± 0.018 |
| Channel Islands | ||
| Fort Doyle, Guernsey | Jan. 1989 | 1.093 ± 0.003 |
| Feb. 1989 | 1.103 ± 0.004 | |
| Mar. 1989 | 1.092 ± 0.006 | |
| Apr. 1989 | 1.098 ± 0.006 | |
| Mean | (1989) | 1.097 ± 0.005 |
| Scotland (urban) | ||
| Edinburgh | Feb. 1990 | 1.096 ± 0.007 |
| May 1990 | 1.069 ± 0.004 | |
| May 1990 | 1.086 ± 0.005 | |
| May 1990 | 1.119 ± 0.001 | |
| May 1990 | 1.088 ± 0.005 | |
| May 1990 | 1.086 ± 0.002 | |
| May 1990 | 1.087 ± 0.002 | |
| June 1990 | 1.090 ± 0.002 | |
| June 1990 | 1.097 ± 0.002 | |
| July 1990 | 1.094 ± 0.002 | |
| Aug. 1990 | 1.090 ± 0.003 | |
| Apr. 1991 | 1.092 ± 0.003 | |
| July 1991 | 1.099 ± 0.019 | |
| Nov. 1991 | 1.101 ± 0.006 | |
| Mean | (1990–1991) | 1.092 ± 0.011 |
| Scotland (urban) | ||
| Glasgow | Jan. 1991 | 1.095 ± 0.003 |
| Apr. 1991 | 1.102 ± 0.004 | |
| July 1991 | 1.097 ± 0.006 | |
| Oct. 1991 | 1.093 ± 0.003 | |
| Jan. 1992 | 1.114 ± 0.005 | |
| July 1992 | 1.096 ± 0.003 | |
| Oct. 1992 | 1.093 ± 0.001 | |
| Mean | (1991–1992) | 1.099 ± 0.007 |
Atmospheric particulates from ∼9 m above street level in the centre of Edinburgh (n = 14) were collected over 8 h sampling periods in 1990–1991 (Table 4) using a membrane filter of pore size 0.45 µm (Millipore, HAWP 037 00) attached to an electric pump (Millipore, XX60 220 50).42 Other atmospheric particulate samples, similarly collected (over 1–2 week periods) in 1985–1986 from the west end of Glasgow (n = 4) and in 1985 from rural locations (except for Devilla, Fife) in northern Scotland (n = 10) and England (n = 5) (Table 4), were also supplied by Dr. Cape. In addition, Mr. D. Smith, National Radiological Protection Board (NRPB), Glasgow, provided large (22 × 25 cm) high-volume air filters collected over 2 week periods in southwest Glasgow (n = 7) in 1991–1992 and in Guernsey (n = 4) in 1989, from which subsamples were cut. All atmospheric filters were leached with 1.6 M nitric acid and the resultant extracts diluted in 2% v/v nitric acid for analysis.
All reagents used in sample preparation were of the highest analytical quality available, i.e. Aristar nitric acid (Merck), and water was deionized and purified by reverse osmosis and ion exchange to 18.3 MΩ quality using a Milli-QSP system (Millipore, Watford, UK).
Sample analysis
All rainwater samples were analysed by ICP-MS using a PlasmaQuad (PQ) 3 instrument (VG Elemental, Winsford, UK), employing an rf forward power of 1350 W, reflected power of 1–3 W, argon gas flows (l min−1) of 13.1, 0.76–0.79 and 0.81–0.85 for coolant, auxiliary and nebulizer, respectively, and nickel sampler and skimmer cones, with a Meinhard nebulizer, Gilson autosampler and a Gilson Minipuls 3 peristaltic pump (Anachem, Luton, UK) providing a solution uptake rate of 0.55 ml min−1. The instrument was operated in peak jumping mode over the mass range 203.6–209.4 u, with one central measurement point per peak, 2 ms dwell time, 1 min acquisition time, five replicate runs per sample and pulse counting mode for ion collection. A solution of the National Institute of Standards and Technology (NIST) common lead isotopic reference standard SRM 981 (206Pb/207Pb = 1.0933) was used for calibration and mass bias correction.42 Over a 1 year period, an average measured value of 1.094 ± 0.002 was obtained.Petrol samples for the periods 1989 to 1991 and 1993 to early 1997 were analysed using VG PQ 1 and 2 instruments, respectively, at the Scottish Universities Research and Reactor Centre (SURRC), East Kilbride, while those for late 1997 to 1998 were analysed by the PQ 3 described above. All pine needle and atmospheric particulate samples, with the exception of those from Edinburgh (1990–1991), were analysed using the VG PQ 2 instrument.
After appropriate blank corrections corresponding to the different modes of preparation, mean internal analytical precisions [1 standard deviation (s)] on the measured 206Pb/207Pb ratios were ±0.24%, ±0.34% and ±0.42% for the petrol (n = 33), rainwater (n = 145) and pine needle/atmospheric particulate (n = 58) samples, respectively. Mean external analytical precision on 206Pb/207Pb, as determined by repeated analysis (n = 55) of the International Atomic Energy Agency SL-1 Lake Sediment reference material over several years, was ±0.36%.
The results presented here are for the 206Pb/207Pb ratio, that most commonly used in environmental studies and the most relevant for the purposes of this research. Partly because of the fragmented nature of this work over several years, a complete set of data for the other lead isotopic ratios is not available.
Results
Petrol
The individual 206Pb/207Pb results (n = 33) for petrol collected in the Edinburgh area from February 1989 to November 1998 are listed in Table 1. The ratios ranged from 1.056 ± 0.001 (February 1989) to 1.098 ± 0.0014 (November 1997) with an overall mean (±1 s) of 1.076 ± 0.011. The mean values (n = 9) for individual periods of collection from 1989 to 1998 are plotted in Fig. 2, displaying an average increase of 0.2% (∼0.002) in 206Pb/207Pb per year over the decade. The mean of 1.065 ± 0.0026 for the October/November 1994 samples is very close to the 1.067 ± 0.007 reported for leaded petrol purchased in Southampton, south England, in November/December 1994.14 | ||
| Fig. 2 Mean (±1 s) 206Pb/207Pb ratios of leaded petrol for various collection periods in Edinburgh from 1989 to 1998 (see Table 1 for details). | ||
Rainwater
Of the 47 rainwater samples collected across Scotland in December 1997 and January 1998, 74% had a lead concentration below 5 µg l−1, with median and maximum concentrations of 2.9 and 29.3 µg l−1, respectively. Of the 68 rainwater samples collected by open gauge at Glensaugh, Hartwood and Sourhope from November 1997 to December 1998, 97% had a lead concentration below 5 µg l−1, with median and maximum concentrations of 1.0 and 8.8 µg l−1, respectively. Corresponding data for the 30 filter gauge samples were 60% < 5 µg l−1, with median and maximum concentrations of 4.1 and 14.9 µg l−1, respectively.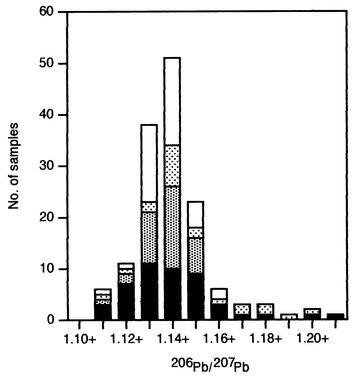 | ||
Fig. 3 Histogram of 206Pb/207Pb ratios for rainwater samples collected at the 25 sampling sites in Scotland during December 1997 and January 1998 (■) and at the three long-term rainwater collection sites, Glensaugh (□), Hartwood ( ) and Sourhope ( ) and Sourhope ( ), from November 1997 to December 1998. ), from November 1997 to December 1998. | ||
For Glensaugh, 90% of all samples collected had 206Pb/207Pb ratios in the range 1.130–1.159, with corresponding figures of 57% for Hartwood and 92% for Sourhope. For the total of 98 samples collected at the three long-term sites, almost 88% of the 206Pb/207Pb ratios were between 1.120 and 1.159, compared with 79% for the Scotland December 1997/January 1998 samples (Fig. 3). Taken together, the complete set of rainwater samples (n = 145) collected at all sites in Scotland from November 1997 to December 1998 had a mean 206Pb/207Pb ratio (±1 s) of 1.144 ± 0.017, with 85% of values lying between 1.120 and 1.159.
Pine needles and atmospheric particulates
For Scotland, pine needles of 1985 growth from rural locations in the north displayed a mean 206Pb/207Pb ratio (±1 s) of 1.113 ± 0.005 (n = 4), indistinguishable from the 1.118 ± 0.006 (n = 10) recorded for atmospheric particulates collected at the predominantly northern rural locations in 1985 (Table 4). In contrast, urban atmospheric particulates collected in Glasgow in 1985 had a lower mean 206Pb/207Pb ratio (±1 s) of 1.085 ± 0.012 (n = 4), a value similar to the 1.099 ± 0.007 (n = 7) found for the same city in 1991–1992 and the 1.092 ± 0.011 (n = 14) reported for urban Edinburgh in 1990–199142 (Table 4), when the mean 206Pb/207Pb ratio of leaded petrol sold in the city was 1.079 ± 0.011 (n = 7) (Table 1).Elsewhere in Britain, the mean 206Pb/207Pb ratio (±1 s) of atmospheric particulates from rural Gisburn in northwest England in 1985 was 1.108 ± 0.018 (n = 5), similar to the corresponding Scottish average and to the values for two 1985 pine needle samples from southeast England (Table 4).
On mainland Europe, the mean 206Pb/207Pb ratio (±1 s) for pine needles from four sites in the Netherlands and Germany from 1982 to 1986 was 1.126 ± 0.006 (n = 8), slightly higher than the overall mean (±1 s) of 1.112 ± 0.010 (n = 21) for the British pine needle and rural atmospheric particulate values for the period 1985–1986 (Table 4). The average 206Pb/207Pb ratio (±1 s) of 1.097 ± 0.005 for atmospheric particulates in the Channel Islands in 1989 was closer to the values observed for atmospheric particulates (n = 25) in urban Glasgow and Edinburgh from 1985 to 1992, when 52% lay between 1.090 and 1.099 and 76% between 1.080 and 1.099 (Table 4).
Discussion
For the samples of this study collected in Scotland, Fig. 4 displays the mean 206Pb/207Pb ratios (±1 s) for petrol (1989–1998, P), urban atmospheric particulates in 1985 (APU1) and in 1990–1992 (APU2), rural atmospheric particulates in 1985 (APR) and rainwater in 1997–1998 (RW2). Also included are the mean 206Pb/207Pb ratios (±1 s) for rainwater (1.120 ± 0.016) collected at the three long-term sites in Scotland in 1989–1991 (RW1)35 and for coal (1.181 ± 0.011) collected at 30 sites in Scotland.43 The strong influence of car exhaust emissions of lead, originating from the comparatively 206Pb-depleted Australian lead (206Pb/207Pb ∼ 1.04) used with British Columbian lead (206Pb/207Pb ∼ 1.16) as the main source3 of alkyllead manufacture for leaded petrol (206Pb/207Pb = 1.076 ± 0.011), upon atmospheric lead in the urban environment is readily seen from the 206Pb/207Pb ratios for APU1 and APU2 of 1.085 ± 0.012 and 1.094 ± 0.010, respectively. That rural environments remote from urban centres are less strongly influenced by such emissions is reflected in the 206Pb/207Pb ratio of 1.118 ± 0.006 for APR compared with 1.085 ± 0.012 for APU1, both for 1985, and by the 206Pb/207Pb ratio of 1.120 ± 0.016 for RW1 (Glensaugh, Hartwood and Sourhope) in 1989–199135 compared with 1.094 ± 0.010 for APU2 in 1990–1992. Similarly, with respect to RW2, the mean 206Pb/207Pb ratio of 1.150 ± 0.008 for rainwater in December 1997/January 1998 from the most remote and rural of the Scottish locations, the north/northwest, was greater than that from the densely populated central belt, viz. 1.131 ± 0.010, excluding the outlier from Stirling for January 1998 (Table 2). | ||
| Fig. 4 Mean (±1 s) 206Pb/207Pb ratios for petrol (1989–1998, P), urban atmospheric particulates (1985, APU1; 1990–1992, APU2), rural atmospheric particulates (1985, APR), rainwater (1989–1991, RW1; 1997–1998, RW2) and coal (C) from Scotland. | ||
An increase in the atmospheric 206Pb/207Pb ratio in Scotland from 1989–1991 to 1997–1998 of ∼0.02–0.03 is indicated by the summary data for Glensaugh, Hartwood and Sourhope and for Scotland overall in Table 5. Using the averages (±1 s) of 1.120 ± 0.016 (n = 31) and 1.144 ± 0.017 (n = 145) based on all individual 1989–1991 and 1997–1998 data, respectively, the statistically significant (p < 0.01, t-test) increase in the atmospheric 206Pb/207Pb ratio is 0.024 ± 0.023. Over the same time period, the 206Pb/207Pb ratio for petrol increased (p < 0.05, t-test) by 0.013 ± 0.015 from 1.075 ± 0.013 (1989–1991) to 1.088 ± 0.007 (late 1997–1998). This suggests that the reduction in the emission of lead to the atmosphere from car exhausts from 2.6 × 103 tonnes in 1989 to 0.8 × 103 tonnes in 1997, when the consumption of unleaded petrol accounted for 71.9% of sales,39 has contributed to the observed increase in the 206Pb/207Pb ratio for rainwater in Scotland.
| 206Pb/207Pb (±1 s) | ||||
|---|---|---|---|---|
| Location | Date of collection | n | Range | Mean |
| Glensaugh | 1989–199135 | 11 | 1.101–1.152 | 1.119 ± 0.013 |
| 1997–1998 (O) | 27 | 1.113–1.163 | 1.140 ± 0.010 | |
| 1997–1998 (F) | 14 | 1.135–1.161 | 1.144 ± 0.008 | |
| Hartwood | 1989–199135 | 11 | 1.104–1.153 | 1.129 ± 0.014 |
| 1997–1998 (O) | 13 | 1.123–1.207 | 1.164 ± 0.024 | |
| 1997–1998 (F) | 8 | 1.118–1.149 | 1.141 ± 0.010 | |
| Sourhope | 1989–199135 | 9 | 1.081–1.132 | 1.110 ± 0.018 |
| 1997–1998 (O) | 28 | 1.112–1.153 | 1.140 ± 0.008 | |
| 1997–1998 (F) | 8 | 1.129–1.155 | 1.147 ± 0.008 | |
| Scotland (Table 2) | Dec. 1997–Jan. 1998 | 47 | 1.112–1.218 | 1.144 ± 0.021 |
| Scotland (all) | 1997–1998 | 145 | 1.112–1.218 | 1.144 ± 0.017 |
Source apportionment calculations using an equation6,44 such as
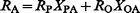 | (1) |
With respect to estimation of the contribution of lead from petrol to the emissions of lead to the atmosphere, the above approach based on the isotopic characterization of petrol lead and atmospheric lead does not take into account the likelihood that the possible uniqueness of the source lead isotopic signature, defined in terms of the geochemical origins of the lead in the parent material, will not be restricted to one particular anthropogenic source in terms of the mode or pathway of release to the atmosphere. For example, the smelting of lead ores used to produce lead for the manufacture of alkyllead additives will release lead to the atmosphere of the same isotopic signature as car exhaust emissions of lead from leaded petrol. For 1994, AEA Technology46 estimated that emissions of lead to the atmosphere in the UK comprised 1295 tonnes from petrol lead, 149 tonnes from waste-related sources, including incineration of sewage, tyres and clinical wastes, 140 tonnes from non-ferrous metal (including lead) production, 102 tonnes from coal (91%) and oil (9%) burning, 46 tonnes from iron and steel production, and 22 tonnes from industrial processes such as cement, glass and coke production. Some of these sources (e.g. waste incineration, recycling and refining of battery lead), which will increase in relative importance47 as emissions of lead from car exhausts continue to fall with increasing consumption of unleaded petrol and the banning of leaded petrol in the UK in 2000, may be of ill-defined or varying lead isotopic signature. Monna et al.,14 however, found a mean 206Pb/207Pb ratio of 1.149 ± 0.005 (range 1.143–1.155) for French urban incinerator ashes in 1993–1994, similar to the 1.142–1.159 found in Germany by Hamester et al.12 The effect of using this value as the predominant “industrial" signature (i.e. as RO) in source apportionment eqn. (1) would be to reduce the calculated contribution of car exhaust emissions of lead to atmospheric lead even further, and there is recent evidence from a New York study48 that municipal waste incineration releases of lead have been seriously underestimated in the past.
The approach adopted in this study does not take into account likely differences, depending upon proximity to major sources, in the atmospheric inventory of lead at different locations. It also neglects the possibility that clouds, as with the movement of aerosol-entraining air masses, can transport lead from much further afield, with the result that wet precipitation may not be just washing out lead released to the atmosphere in the geographical area where the rainwater samples are collected. Whether or not the slightly elevated mean 206Pb/207Pb ratio of 1.162 ± 0.020 for rainwater in eastern Scotland in December 1997/January 1998, supported to some extent by the average Glensaugh/Sourhope 206Pb/207Pb ratio of 1.149 ± 0.008 for that period, could reflect the influence of air masses from the continent of Europe to the east is difficult to assess, but typical 206Pb/207Pb ratios of 1.148–1.162 and 1.160–1.185 have been reported for air masses from the regions of USSR/Finland and Eastern Europe,4 respectively, and 206Pb/207Pb ratios ranging from 1.109 up to 1.204 were observed in aerosols above Helgoland in the southeastern North Sea during a 2 week period in April 1988.45 More recently in Western Europe, however, it is worth noting that there was a shift in the mean 206Pb/207Pb ratio of aerosols collected from a rural area in the Nord-Pas de Calais region along the English Channel from 1.108 ± 0.005 in 1982–1983 to 1.148 ± 0.003 in 1994, paralleled by similar variations from 1.115 ± 0.008 in 1981–1989 to 1.143 ± 0.006 in 1992–1995 in urban aerosols in France and from 1.094 in 1985 to 1.135 ± 0.002 in 1995 along French highways.33 These increases from values similar to those found in our study for pine needles in Western European countries and for atmospheric particulates in the Channel Islands during the 1980s (Table 4) are analogous to those observed here for Scotland since the 1980s and can also be attributed to the declining influence of lead emissions as a consequence of the phasing out of leaded petrol in France.
Conclusions
The 206Pb/207Pb ratio of rainwater in Scotland has increased during the 1990s from 1.120 ± 0.016 in 1989–1991 to 1.144 ± 0.017 in 1997–1998, attributable partially to an increase in the 206Pb/207Pb ratio of leaded petrol, but mainly to a reduction in the car exhaust emissions of lead to the atmosphere resulting from the increased consumption of unleaded petrol and the phasing out of alkyllead additives. In view of the wide ranging use made of anthropogenic lead isotopic ratios as tracers and source indicators in environmental and earth sciences, it will be important to continue monitoring atmospheric 206Pb/207Pb ratios at this time of change and to identify contributions of lead from sources, such as incinerators, which may increase in relative importance.Acknowledgements
We thank J. N. Cape, NERC ITE, and D. Smith, NRPB, for provision of certain samples, C. L. Sugden, K. Cotton, J. L. Hitchen, A. A. Pott, C. F. Catto, M. Emery and N. Kulasuriya for assistance with collection and preparation of most samples, and A. B. MacKenzie, M. McCartney, T. M. Shimmield and K. Sampson for those ICP-MS analyses carried out using the VG PQ 1 and 2 at SURRC, East Kilbride.References
- S. Moorbath, Philos. Trans. R. Soc. London, Ser. A, 1962, 254, 295 Search PubMed.
- T. J. Chow, in Isotope Ratios as Pollutant Source and Behaviour Indicators, IAEA, Vienna, 1975, pp. 95–107. Search PubMed.
- H. T. Delves, Chem. Brit., 1988, 24, 1009 Search PubMed.
- J. F. Hopper, H. B. Ross, W. T. Sturges and L. A. Barrie, Tellus, 1991, 43B, 45 Search PubMed.
- M. Keinonen, Sci. Total Environ., 1992, 113, 251 CrossRef CAS.
- J. G. Farmer, C. L. Sugden, A. B. MacKenzie, G. H. Moody and M. Fulton, Environ. Technol., 1994, 15, 593 Search PubMed.
- W. T. Sturges and L. A. Barrie, Atmos. Environ., 1989, 23, 1645 CrossRef CAS.
- W. T. Sturges and L. A. Barrie, Atmos. Environ., 1989, 23, 2513 CrossRef CAS.
- H. Mukai, N. Furuta, T. Fujii, Y. Ambe, K. Sakamoto and Y. Hashimoto, Environ. Sci. Technol., 1993, 27, 1347 CAS.
- W. T. Sturges, J. F. Hopper, L. A. Barrie and R. C. Schnell, Atmos. Environ., 1993, 27A, 2865 CAS.
- F. E. Grousset, C. R. Quetel, B. Thomas, P. Buat-Menard, O. F. X. Donard and A. Bucher, Environ. Sci. Technol., 1994, 28, 1605 CAS.
- M. Hamester, H. Stechmann, M. Steiger and W. Dannecker, Sci. Total Environ., 1994, 146/147, 321 CrossRef CAS.
- M. Chiaradia, B. L. Gulson, M. James, C. W. Jameson and D. Johnston, Atmos. Environ., 1997, 31, 3511 CrossRef CAS.
- F. Monna, J. Lancelot, I. W. Croudace, A. B. Cundy and J. T. Lewis, Environ. Sci. Technol., 1997, 31, 2277 CrossRef CAS.
- J. R. Graney, A. N. Halliday, G. J. Keeler, J. O. Nriagu, J. A. Robbins and S. A. Norton, Geochim. Cosmochim. Acta, 1995, 59, 1715 CrossRef CAS.
- J. G. Farmer, L. J. Eades, A. B. MacKenzie, A. Kirika and A. E. Bailey-Watts, Environ. Sci. Technol., 1996, 30, 3080 CrossRef CAS.
- J. G. Farmer, A. B. MacKenzie, C. L. Sugden, P. J. Edgar and L. J. Eades, Water, Air, Soil Pollut., 1997, 100, 253 CrossRef CAS.
- W. Shotyk, D. Weiss, P. G. Appleby, A. K. Cheburkin, R. Frei, M. Gloor, J. D. Kramers, S. Reese and W. O. van der Knaap, Science, 1998, 281, 1635 CrossRef CAS.
- C. E. Dunlap, E. Steinnes and A. R. Flegal, Earth Planet. Sci. Lett., 1999, 167, 81 CrossRef CAS.
- D. Weiss, W. Shotyk, P. G. Appleby, J. D. Kramers and A. K. Cheburkin, Environ. Sci. Technol., 1999, 33, 1340 CrossRef CAS.
- J. R. Bacon, M. L. Berrow and C. A. Shand, Int. J. Environ. Anal. Chem., 1992, 46, 71 Search PubMed.
- J. R. Bacon, M. L. Berrow and C. A. Shand, Int. J. Environ. Anal. Chem., 1995, 59, 253 Search PubMed.
- N. Walraven, B. J. H. van Os, G. Th. Claver, J. H. Baker and S. P. Vriend, J. Geochem. Explor., 1997, 59, 47 CrossRef CAS.
- J. R. Bacon, K. C. Jones, S. P. McGrath and A. E. Johnston, Environ. Sci. Technol., 1996, 30, 2511 CrossRef CAS.
- S. A. Watmough, R. J. Hughes and T. C. Hutchinson, Environ. Sci. Technol., 1999, 33, 670 CrossRef CAS.
- G. Aberg, J. M. Pacyna, H. Stray and B. L. Skjelkvale, Atmos. Environ., 1999, 33, 3335 CrossRef.
- K. J. Rosman, C. Ly and E. Steinnes, Environ. Sci. Technol., 1998, 32, 2542 CrossRef CAS.
- D. Weiss, W. Shotyk, J. D. Kramers and M. Gloor, Atmos. Environ., 1999, 33, 3751 CrossRef CAS.
- K. J. R. Rosman, W. Chisholm, C. F. Boutron, J.-P. Candelone and U. Gorlach, Nature, 1993, 362, 333 CrossRef CAS.
- K. J. R. Rosman, W. Chisholm, S. Hong, J.-P. Candelone and C. F. Boutron, Environ. Sci. Technol., 1997, 31, 3413 CrossRef CAS.
- H. Maring, D. M. Settle, P. Buat-Menard, F. Dulac and C. C. Patterson, Nature, 1997, 300, 154.
- A. Veron and T. M. Church, J. Geophys. Res., 1997, 102, 28049 CAS.
- A. Veron, P. Flament, M. L. Bertho, L. Alleman, R. Flegal and B. Hamelin, Atmos. Environ., 1999, 33, 3377 CrossRef CAS.
- G. T. Shen and E. A. Boyle, J. Geophys. Res., 1988, 93, 15715.
- J. R. Bacon and D. C. Bain, Environ. Geochem. Health, 1995, 17, 39 Search PubMed.
- Y. Erel, A. Veron and L. Halicz, Geochim. Cosmochim. Acta, 1997, 61, 4495 CrossRef CAS.
- Y. Gelinas and J.-P. Schmit, Environ. Sci. Technol., 1997, 31, 1968 CrossRef CAS.
- A. B. MacKenzie, J. G. Farmer and C. L. Sugden, Sci. Total Environ., 1997, 203, 115 CrossRef CAS.
- DETR, Digest of Environmental Statistics No. 20, The Stationery Office, London, 1998. Search PubMed.
- A. Cocherie, P. Negrel, S. Roy and C. Guerrot, J. Anal. At. Spectrom., 1998, 13, 1069 RSC.
- W. J. Price, Spectrochemical Analysis by Atomic Absorption, Heyden and Son Ltd., London, 1979, p. 240. Search PubMed.
- C. L. Sugden, J. G. Farmer and A. B. MacKenzie, Environ. Geochem. Health, 1993, 15, 59 Search PubMed.
- J. G. Farmer, L. J. Eades and M. C. Graham, Environ. Geochem. Health, 1999, 21, 257 Search PubMed.
- H. T. Delves and M. J. Campbell, Environ. Geochem. Health, 1993, 15, 75 Search PubMed.
- M. KerstenU. ForstnerP. KrauseM. KriewsW. DanneckerC.-D. Garbe-SchonbergM. HockU. TerzenbachH. Graßl, in Impact of Heavy Metals in the Environment, ed. J.-P. Vernet, Elsevier, Amsterdam, 1992, vol. 2, pp. 311–325. Search PubMed.
- AEA Technology, http://www.aeat.co.uk/netcen/airqual/emissions/index/html, 1998..
- J. O. Nriagu, Science, 1998, 281, 1622 CrossRef CAS.
- S. N. Chillrud, R. F. Bopp, J. H. Simpson, J. M. Ross, E. L. Shuster, D. A. Chaky, D. C. Walsh, C. C. Choy, L.-R. Tolley and A. Yarme, Environ. Sci. Technol., 1999, 33, 657 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |