Simultaneous monitoring of atmospheric methane and speciated non-methane hydrocarbon concentrations using Peltier effect sub-ambient pre-concentration and gas chromatography
David
Harrison
a,
Paul W.
Seakins
*a and
Alastair C.
Lewis
*b
aSchool of Chemistry, University of Leeds, Leeds, UK LS2 9JT. E-mail: p.w.seakins@chem.leeds.ac.uk
bSchool of Chemistry and School of the Environment, University of Leeds, Leeds, UK LS2 9JT. E-mail: a.c.lewis@chem.leeds.ac.uk
First published on 1st February 2000
Abstract
Sub-ambient trapping, used to pre-concentrate atmospheric samples for non-methane hydrocarbon (NMHC) analysis by gas chromatography, can also be used to measure ambient methane concentrations. Above a sample volume of 40 ml, a dynamic equilibrium is established between ambient and trapped methane allowing for simultaneous quantitative determinations of methane and NMHC. The temperature stability of the trap is critical for quantitative methane analysis and this can be achieved by Peltier effect cooling. Simultaneous measurements of methane and NMHC reduce the equipment required for field trips and can ease the interpretation and modelling of atmospheric data. The feasibility for deployment of the system in remote locations was demonstrated by running the apparatus virtually unattended for a 5-day period. The correlations between the concentrations of methane, ethane and ethene measured during this period are discussed.
Introduction
Methane, the simplest hydrocarbon, is present in the atmosphere at much greater background concentrations (typically, approximately 1.8 parts per million by volume, ppmv)1 than other saturated or unsaturated hydrocarbon species (non-methane hydrocarbons, NMHCs). Concentrations of these latter compounds can range from parts per billion by volume (ppbv) levels in urban environments2 to sub parts per trillion by volume (pptv) levels in remote locations.3 Methane is conventionally monitored using specific instruments based on either gas chromatographic4 or infrared spectroscopic5 methods.However, in many cases, atmospheric modellers are interested in simultaneous determinations of methane and speciated NMHCs. For example, methane and total NMHCs can both contribute at a similar level to the removal of OH radicals at the remote monitoring site at Mace Head, Eire.6 Modelling of OH removal, essential to our understanding of this crucial atmospheric species, is only possible with a knowledge of simultaneous measurements of speciated NMHCs and methane. Speciation is required as each hydrocarbon will react at a different rate with OH radicals. Apparatus capable of making simultaneous methane and NMHC determinations would facilitate modelling (as measurements are made simultaneously from the same location, there is no need to interpolate concentrations) and minimize the experimental equipment required. Additionally, methane has a different source profile from other hydrocarbons and hence simultaneous methane and NMHC determinations yield more information on the chemical history of an air mass.
In this paper, we outline a method for making simultaneous measurements of methane and NMHCs based on gas chromatographic techniques developed previously in this laboratory.7–9 The method is shown to give a linear response over the range of methane typically found in the atmosphere. A previous publication from this department10 showed that methane and NMHCs could be determined using basically the same apparatus, however, unlike the measurements reported in this paper, no simultaneous measurements were performed, methane being loop injected directly into the column with NMHCs being concentrated using a sorbent tube prior to rapid desorption into the column. The feasibility of the method for determining methane and NMHC concentrations in remote locations is demonstrated by running the instrument virtually unattended over an extended period.
Experimental
A full description of the apparatus can be found elsewhere.7–9 In this paper, we include a brief summary of the apparatus and focus on the development and calibrations required for the simultaneous determination of methane and higher molecular weight hydrocarbons.Ambient air is drawn through a sorbent tube packed with 80 mg of activated coconut charcoal (PhaseSep, Deeside, UK) and cooled to sub-ambient levels (−15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C) using either liquid release CO2 or Peltier plates (Fig. 1A,B). After collection of a suitable sample (∼1 l of air), the sorbent tube is rapidly heated (−15 to 400°C at 16°C s−1) using a programmed temperature vaporization injector (PTV, Optic 400, Ai, Cambridge, UK) and swept onto the analytical column by a flow of helium carrier gas (27 ml min−1). A thermocouple located on the sorbent tube is linked directly to the PTV system. Sample separation is achieved using an Al2O3 PLOT column (Na2SO4 stabilized, 50 m, 0.53 mm id, Chrompack, Bergen op Zoom, The Netherlands) capable of separating C1–C9 hydrocarbons with detection by flame ionization. The column is housed within a programmable chromatographic oven (Ai GC94, Ai, Cambridge, UK).
°C) using either liquid release CO2 or Peltier plates (Fig. 1A,B). After collection of a suitable sample (∼1 l of air), the sorbent tube is rapidly heated (−15 to 400°C at 16°C s−1) using a programmed temperature vaporization injector (PTV, Optic 400, Ai, Cambridge, UK) and swept onto the analytical column by a flow of helium carrier gas (27 ml min−1). A thermocouple located on the sorbent tube is linked directly to the PTV system. Sample separation is achieved using an Al2O3 PLOT column (Na2SO4 stabilized, 50 m, 0.53 mm id, Chrompack, Bergen op Zoom, The Netherlands) capable of separating C1–C9 hydrocarbons with detection by flame ionization. The column is housed within a programmable chromatographic oven (Ai GC94, Ai, Cambridge, UK).
 | ||
| Fig. 1 Schematic diagrams of the CO2 (A) and Peltier (B) cooled injection systems. | ||
Initial experiments utilized liquid release CO2 to cool the sorbent trap with the flow of CO2 being controlled by a solenoid valve actuated via a signal from the PTV system linked to a feedback thermocouple mounted on the trap (Fig. 1A). Although relatively simple in design, this system has a number of drawbacks making it unsuitable for methane determinations in remote locations. Firstly, the CO2 cylinders need regular replacement and are impractical to take on field campaigns. Secondly, and more importantly, temperature stability depends critically on the CO2 flow rate and can fluctuate by several degrees. As we shall demonstrate in the following section, such small-scale temperature fluctuations are unimportant for quantitative NMHC determinations, but trap temperature stability is critical when an equilibrium reconcentration approach is applied to the simultaneous determination of methane.
The PTV and adsorbent trap system were subsequently modified to utilize Peltier effect cooling to replace CO2 (Fig. 1B). Two 50 W two stage Peltier devices (RS, UK) were used, powered by a 12 V dc power supply unit at 12 A. The cold sides of the Peltier plates are thermally bonded to a metal ‘cold' block that surrounds the external housing of the sorbent trap using zinc impregnated silicone grease. At the hot sides (ΔT = 80°C) of the Peltier plates heat is removed via liquid flow cells flushed with ethylene glycol maintained at −8°C by a chiller (Grant L30). The Peltier and chiller system is run continuously. Since the ‘cold' block is essentially detached from the PTV injector, the presence of Peltier cooling makes little difference to the rate of heating observed during the injection process. Continuous operation gives greater stability and simplifies the system. The large thermal mass of the heat sink block allows the thermocouple feedback of the injector to control the adsorbent temperature with a precision of ±0.1°C. Fig. 2 shows that the trap cools down to the operating trapping temperature of −15°C in 280 s and can, in this form, reach an ultimate temperature of −30°C.
 | ||
| Fig. 2 Cooling characteristics of the sub-ambient adsorbent trap cooled with Peltier plates. | ||
Peak resolution and retention time on a PLOT column can be affected by the presence of excess water in the sample. For the experiments described in this paper, ambient water vapour was removed by passing the air through a potassium carbonate trap. This lead to stable retention times, but the trap required daily changing. Subsequently, we have used a Dreschel flask immersed in the chiller to remove most of the water, with the final traces being taken up by either a potassium carbonate or magnesium perchlorate trap. With the Dreschel flask in operation, the solid traps can be left for over a month before needing to be replaced.3
Breakthrough volumes and calibration
Fig. 3 shows a typical urban air chromatogram obtained with the Peltier effect cooled trap. Despite the dramatically different concentrations of ambient methane and NMHCs, the peak heights of methane and the more prominent NMHCs are similar, reflecting that considerable sample breakthrough has occurred for methane. Despite breakthrough we shall demonstrate that we can still obtain a quantitative analysis of methane. In fact, methane breakthrough, is essential for the accurate analysis of lower molecular weight NMHCs, which would otherwise be swamped by a large methane peak. | ||
| Fig. 3
A typical chromatogram of methane and NMHCs obtained using a Peltier cooled injection system (column isothermal at 50°C for 3 min, then 6°C min−1 to 195°C which was maintained for a further 6 min). Inset: chromatogram obtained during continuous operation at the University of Leeds (column maintained isothermal at 50°C). Key: 1, air peak; 2, methane; 3, ethane; 4, ethene; 5, propane; 6, propene; 7, 2-methylpropane; 8, n-butane; 9, acetylene; 10, trans-2-butene; 11, butene; 12, iso-but-1-ene; 13, cis-2-butene; 14, 2-methylbutane; 15, n-pentane; 16, 1,3-butadiene; 17, pentenes; 18, 2-methylpentane; 19, 3-methylpentane; 20, n-hexane; 21, methylhexanes and methylhexenes; 22, heptane; 23, methylcyclopentane; 24, Benzene; 25, toluene.
| ||
Sample breakthrough occurs when an analyte entering the sub-ambient trap migrates entirely through the sorbent bed, eventually eluting from the trap, giving an underestimation of the true sample concentration.11–13 The effect is analogous to that of retention volume on a packed gas chromatograph column. The breakthrough volume for an analyte is not dependent on the concentration, assuming that the sample capacity of the trap is not exceeded. Breakthrough volumes depend on the affinity of the analyte to the sorbent material and, for charcoal, breakthrough is essentially a function of boiling point and hence molecular weight for a given homologous series. Fig. 4 shows the effect of break through for the C2–C6 alkanes. Ethane, the most volatile compound, begins to break through at ∼300 ml at a trap temperature of 15°C, well below the volume required for sufficient sensitivity in remote locations. However, at –15°C, the breakthrough volume for ethane has increased to 1300 ml, sufficient for analysis at the sub ppbv levels present in unpolluted atmospheres.3 As an alternative to cooling, breakthrough volumes can be extended by increasing the mass of adsorbent in the trap, however, this will result in a wider band of analytes introduced to the capillary column upon injection, hence lowering the chromatographic resolution.
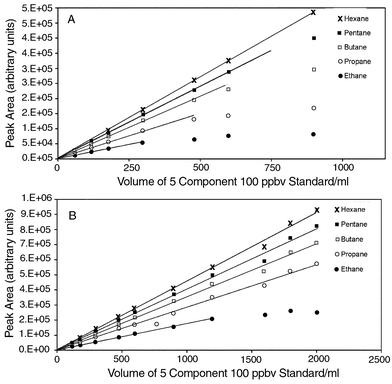 | ||
| Fig. 4
Peak area vs. sample volume for a C2–C6 n-alkane standard at 15°C (A) and −17°C (B). Standard samples were drawn through an activated charcoal trap at 100 ml min−1 for a period corresponding to the required sample volume. Standards were prepared by diluting a 50 ppmv C1–C6 n-alkane standard (Phase Separations, Deeside, UK) with zero nitrogen (BOC, UK). During the analysis, the column was maintained at 50°C for 3 min, then ramped at 6°C min−1 to 195°C and maintained at 195°C for a further 6 min.
| ||
The breakthrough experiments depicted in Fig. 4 were repeated for methane. The results are shown in Fig. 5. No methane is seen for the first 4 ml of the sample, corresponding to the dead volume of the sampling and injector system. Thereafter, there is a linear increment of detector signal with sample volume as sample is loaded without breakthrough. After approximately 10 ml of sample, the plots begin to curve as the methane starts to break through (significantly smaller breakthrough volume than for ethane reflecting the reduced affinity of the trap for methane). Above 20 ml of sample, there is no increase in signal, i.e. a steady state exists between loading and breakthrough. Provided that the sample volume is greater than 20 ml, the peak area will always be the same for a constant methane concentration.
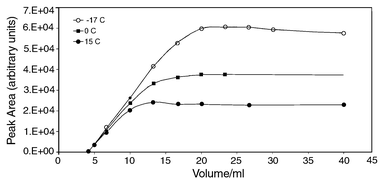 | ||
| Fig. 5
Peak area vs. sample volume for a methane standard (1.3 ppmv in N2, BOC, Leeds, UK) at −15, 0 and 15°C. Standard samples were drawn through an activated charcoal trap at 10 ml min−1 for a variable time period corresponding to the required sample volume. Analysis was performed with the PLOT column held isothermally at 50°C.
| ||
As stated above, breakthrough depends on the sample volume and not on the sample concentration. Once a steady state has been obtained between loading and breakthrough, the observed signal will depend on the capacity of the trap (dependent on the temperature of the trap) and the concentration of the sample. A dynamic equilibrium reconcentration is therefore achieved. The response of the system to varying sample concentrations is shown in Fig. 6. Linearity is excellent over the range of methane concentrations that would be generally encountered in the urban or rural background atmospheres. The varying methane concentrations in Fig. 6 were calculated from the known flow rates of either a 50 or 1 ppmv methane standard and diluent and verified using a separate loop injection methane GC instrument.14
 | ||
| Fig. 6
Methane peak area vs. methane concentration with high (A) and low (B) methane concentrations. Variable concentrations were obtained by diluting 50 ppmv (Phase Separations, UK) (A) or 1.016 ppmv (B) methane standards with a continual stream of synthetic air (Air Products, Crewe, UK). The samples were drawn through an activated charcoal trap at 100 ml min−1 for 2 min. Analysis was performed with the PLOT column held isothermally at 50°C. Error bars represent one standard deviation. The two determinations in (B) refer to methane concentrations referenced either by calculated flow rates or measurements on a loop injector GC.
| ||
Competitive adsorption is an issue which needs to be addressed in such studies. The capacity of the trap will be determined by the co-adsorption of other compounds within the sample. This is a difficult area to address without using the trap as a real column and then seeing whether the eluting peaks are non-Gaussian in nature. However, we are confident that competitive adsorption will not be an issue for normal atmospheric samples for the following two reasons.
(1) Firstly, NMHC calibrations for subsequent campaigns2 have used both artificial and real ppbv air samples as calibrant mixtures. There has been no evidence of competitive adsorption in either case for sample loadings considerably greater than those used in this study.
(2) Secondly, the capacity of the trap greatly exceeds that of the PLOT column used in these studies (capacity ∝ diameter5/2, diameter of column = 0.53 mm, diameter of trap = 4 mm, giving the trap a capacity greater than the column by a factor of 156). The peaks eluting from the column are always Gaussian under atmospheric conditions, indicating that the column is not overloaded and hence neither is the trap.
The reproducibility of the system for methane determinations is excellent. Repeated analysis of a (17.9 ppmv) methane standard (seven samples) gave 3.8% (one standard deviation) for the CO2 cooled trap and just 0.7% for the Peltier system. The longer term stability of the Peltier system was demonstrated by the 1.4% standard deviation for eight samples taken at the beginning and end of a 5-day period. The efficiency of the CO2 cooled trap was dependent on the CO2 flow characteristics which can change over the course of several days due to movement of the nozzle or level of the CO2 in the cylinder. Frequent calibrations were therefore required for the CO2 cooled system.
Continuous sampling
The ability of the Peltier cooled apparatus for obtaining continuous unattended measurements was demonstrated by a 5-day campaign based at the University of Leeds. The system was deployed to obtain continuous samples of methane, ethane and ethene at a 12 min frequency from a courtyard outside the School of Chemistry. To obtain this sampling frequency, the oven temperature was operated in an isothermal mode at 50°C. A typical chromatogram is shown in the inset of Fig. 3.
The results are shown in Fig. 7. Methane concentrations peak at around 5 ppmv, which is high compared with previously published methane concentrations for urban areas;15–18 however, the location adjacent to the School of Chemistry is not typical of the urban background as a whole. Methane concentrations approached, but never fell below, the value of 1.8 ppmv which is considered to be the background level for the Northern Hemisphere.1
 | ||
| Fig. 7 Ambient concentrations (ppmv/ppbv) of methane, ethane and ethene measured with a 12 min sampling frequency over a 5-day period. | ||
The primary urban source of methane is from natural gas leakage, which is also a major source of ethane (56% natural gas leakage, 16% road transport2); therefore we would expect to see a strong correlation between methane and ethane concentrations and indeed this was observed (Fig. 8). The major source of ethene is road transport2 (85%), and therefore the correlation between methane and ethene concentrations is not expected to be as high, consistent with the experimental observations shown in Fig. 8. Although originating from different sources, chemical removal and physical dispersion mechanisms will be similar for all three hydrocarbons and therefore it is not surprising that some degree of correlation between methane and ethene concentrations exists.

Conclusions
Despite the factor of 100 difference in methane and NMHC concentrations, sub-ambient adsorbent trapping with gas chromatographic analysis can be used to accurately monitor both methane and higher speciated NMHCs. Peltier effect cooling is essential for providing the stable trap temperatures required for reproducible methane determinations. It has been demonstrated that the analytical system can be used to obtain unattended, continuous samples over a period of several days.References
- N. A. K. Khalil, R. A. Rasmussen and F. Moreaes, J. Geophys. Res., 1993, 98, 14753.
- Ozone in the United Kingdom, Fourth Report of the Photochemical Oxidants Review Group, Department of the Environment, Transport and the Regions, London, 1977. Search PubMed.
- J. B. McQuaid, A. C. Lewis, K. D. Bartle and S. J. Walton, J. High Res. Chromatogr., 1998, 21, 181 Search PubMed.
- M. C. Shipham, P. M. Crill, K. B. Bartlett, A. H. Goldstein, P. M. Czepiel, R. C. Harriss and D. Blaha, J. Geophys. Res., 1998, 103, 21985 CrossRef.
- D. Brassington, in Transport and Chemical Transformation of Pollutants in the Troposphere, Volume 8: Instrument Development for Atmospheric Research and Monitoring, ed. J. Bosenberg, D. Brassington and P. C. Simon, Springer-Verlag, Berlin, 1997. Search PubMed.
- N. Carslaw D. J. Creasey D. E. Heard A. C. Lewis J. B. McQuaid M. J. Pilling P. S. Monks B. J. Bandy S. A. Penkett, J. Geophys. Res., in the press..
- A. C. Lewis, P. W. Seakins, A. M. Denha, K. D. Bartle and M. J. Pilling, Atmos Environ., 1995, 29, 1871 CrossRef CAS.
- A. C. Lewis, K. D. Bartle, J. B. McQuaid, M. J. Pilling, P. W. Seakins and P. Ridgeon, J. High Res. Chromatogr., 1996, 19, 686 Search PubMed.
- A. C. Lewis, K. D. Bartle, D. E. Heard, J. B. McQuaid, M. J. Pilling and P. W. Seakins, J. Chem. Soc., Faraday Trans., 1997, 93, 2921 RSC.
- A. M. Denha, K. D. Bartle and M. J. Pilling, Anal. Commun., 1994, 31, 321 Search PubMed.
- V. Simon, M. L. Riba, A. Waldhart and L. Torres, J. Chromatogr. A, 1995, 704, 465 CrossRef CAS.
- M. L. Riba, B. Clement, M. Haziza and L. Torres, Toxicol. Environ. Chem., 1991, 31–2, 235 Search PubMed.
- R. J. B. Peters and H. A. Bakkern, Analyst, 1994, 119, 71 RSC.
- D. Harrison, PhD Thesis, University of Leeds, UK, 1999..
- D. R. Blake, V. H. Woo, S. C. Tyler and F. S. Rowland, Geophys. Res. Lett., 1984, 11, 1211 CAS.
- J. H. Shorter, J. B. McManus, C. E. Kolb, E. J. Allwine, B. K. Lamb, B. W. Mosher, R. C. Harriss, U. Partchatka, H. Fischer, G. W. Harris, P. J. Crutzen and H. J. Karbach, J. Atmos. Chem., 1996, 24, 121 CrossRef CAS.
- F. S. Rowland, N. R. P. Harris and D. R. Blake, Nature, 1990, 347, 432 CrossRef.
- M. Aikawa, F. Aotsuka, K. Yoshikawa, M. Tomida, T. Takeuchi and H. Haraguchi, Anal. Sci., 1995, 11, 349 Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |