Competitive binding of the anticancer drug titanocene dichloride to N,N![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) ′-ethylenebis(o-hydroxyphenylglycine) and adenosine triphosphate: a model for TiIV uptake and release by transferrin
′-ethylenebis(o-hydroxyphenylglycine) and adenosine triphosphate: a model for TiIV uptake and release by transferrin
Maolin
Guo
and
Peter J.
Sadler
*
The Department of Chemistry, University of Edinburgh, King’s Buildings, West Mains Road, Edinburgh, UK EH9 3JJ. E-mail: P.J.Sadler@ed.ac.uk
First published on 24th December 1999
Abstract
1H and 31P NMR studies show that in aqueous solution the anticancer agent titanocene dichloride (Cp2TiCl2) binds selectively to N,N![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ′-ethylenebis(o-hydroxyphenylglycine) (H4ehpg) at neutral pH, but preferentially to adenosine triphosphate (ATP) at pH* values below 5.1; intermolecular TiIV transfer from [TiIV(ehpg)(H2O)] to ATP occurs at acidic pH values.
′-ethylenebis(o-hydroxyphenylglycine) (H4ehpg) at neutral pH, but preferentially to adenosine triphosphate (ATP) at pH* values below 5.1; intermolecular TiIV transfer from [TiIV(ehpg)(H2O)] to ATP occurs at acidic pH values.
TiIV complexes are of current medicinal interest owing to the pronounced antitumour properties and low toxic side-effects of two TiIV complexes, titanocene dichloride (Cp2TiCl2) and Budotitane [Ti(bzac)2(OEt)2] (Hbzac = 1-phenylbutane-1,3-dione), which are currently on phase II clinical trials.1,2 Cp2TiCl2 also exhibits pronounced antiviral, antiinflammatory and insecticidal activities.1 Some TiIV complexes have recently been shown to exhibit antibacterial activity.3,4 The significant effectiveness of Cp2TiCl2 against cisplatin-resistant tumour cell lines indicates that it has a different mechanism of action to cisplatin.2 However, in contrast to platinum-based anticancer drugs,5 very little is known about the biological chemistry of titanium and its mechanism of action as an anticancer agent is poorly understood.6 Attack on cellular nucleic acids is believed to be a key process for the antitumour activity of Cp2TiCl2, which inhibits DNA synthesis rather than RNA and protein synthesis, and titanium accumulates in nucleic-acid-rich regions in tumour cells after in vivo or in vitro administration.7,8 However, unlike cisplatin, TiIV does not bind strongly to DNA bases at physiological pH, but forms strong complexes with nucleotides only at pH values below 5.9 Also, there is no evidence for stable complexes of the V or Mo analogues with nucleotides or DNA under physiological conditions.6,10 This raises doubts that nucleic acids are the major target.10,11 Efforts to identify the active TiIV species in biological media have been largely unsuccessful due to the rapid hydrolysis of TiIV complexes at neutral pH and precipitation of inactive polymeric hydrolysis products.12
Recently we found that TiIV–citrate and Cp2TiCl2 bind strongly to the specific FeIII sites of human serum transferrin (hTF) and that TiIV is released from Ti2–hTF at low pH, which may provide a transport mechanism for TiIV.13,14 Transferrin is the 80 kDa iron-transport protein in the blood serum of vertebrates present at a concentration of about 35 μM.15 It takes up FeIII from blood plasma at pH 7.4 and delivers it to cells via receptor-mediated endocytosis. FeIII is released from transferrin in cell compartments called endosomes at pH ca. 5.5.15 The metal binding properties of transferrin have long been mimicked by using the chelating agent N,N′-ethylenebis(o-hydroxyphenylglycine) (H4ehpg).16 This ligand contains donor groups similar to the metal binding sites of transferrin (2Tyr, His, Asp and CO32−). In particular the two tyrosinate ligands are thought to play a dominant role in determining the strength of metal binding to transferrin.17 Recently we found that, in contrast to its rapid hydrolysis at neutral pH in the absence of chelator ligand, Cp2TiCl2 reacts readily with H4ehpg (rac) at pH* 7 and forms a seven-coordinate TiIV complex [Ti(ehpg)(H2O)] (1).18 We have investigated the pH-dependent competitive binding of Cp2TiCl2 to H4ehpg and ATP and the intermolecular transfer of TiIV from the EHPG complex to ATP at low pH. ATP is a potential acceptor ligand for metals released from transferrin in cells and also a purine nucleotide present in DNA (as a phosphate monoester). Our results suggest that novel routes could exist for the transfer of TiIV onto DNA in vivo.
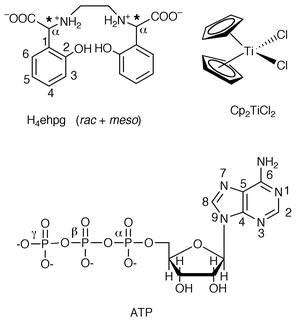
First we carried out solution 1H and 31P{1H} NMR experiments to probe the competitive reaction of Cp2TiCl2(aq) with H4ehpg (rac + meso, ca. 1∶1) and ATP (1∶1∶1 mol ratio, 5 mM), at different pH*† values in D2O. The 1H NMR spectra (purine region and methylene region) and 31P{1H} NMR spectra recorded after 6.5 h of reaction at 298 K are shown in Fig. 1. At pH* 7.0, peaks due to free EHPG‡ [δ 3.12 and 3.03, methylene] and bound Cp ligand [δ 6.42] nearly disappeared, and new peaks characteristic of TiIV–EHPG complexes [δ 2.62 (d) and 2.97 (d), –CH2CH2–; δ 6.72 (d), 7.05 (t) and 7.40 (m), phenyl ring] appeared in the 1H NMR spectrum. None of the ATP reacted, as indicated by both the 1H and 31P NMR spectra. At pH* = 5.1, peaks for both free H4ehpg and free ATP decreased in intensity and new peaks characteristic of TiIV–EHPG and TiIV–ATP complexes [upfield shifted broad peaks for H8 and H2 at δ 8.43 and 8.20, respectively; broad 31P{1H} peak at δ
−24.43 for β phosphate, broad shoulder at δ
−10.85 for α and/or γ phosphate] emerged in both 1H and 31P{1H} NMR spectra. Integration of 1H NMR peaks indicated that ca. 30% of the Cp2TiCl2(aq) formed complexes with EHPG and about 36% of Cp2TiCl2(aq) formed complexes with ATP. However, at pH* ≤ 4.6, the free peaks for EHPG remained unchanged and no peaks for TiIV-bound EHPG were detected, suggesting that TiIV–EHPG complexes were not formed. In contrast, new peaks assignable to TiIV-bound ATP dominated both the 1H and 31P NMR spectra, e.g. the broad upfield-shifted H8, H2 peaks for ATP, and new upfield-shifted broad peaks for the α, β and γ phosphates of ATP. This indicates that Cp2TiCl2(aq) forms complexes with ATP only at these lower pH* values. The broad upfield shifts of H8 and H2 suggest coordination of TiIV to N719 (downfield shifts of H8 and H2 are more common for Pt–N7 coordination, however, upfield shifts have been established for Cp2Mo–N7 coordination19), and the upfield shifts of the new 31P peaks indicate phosphate coordination.
 | ||
| Fig. 1 (A) 1H and (B) 31P{1H} NMR spectra of reactions of Cp2TiCl2(aq), EHPG and ATP (5 mM) in D2O at different pH* values, 298 K, after 6.5 h, showing that Ti–EHPG complexes are formed at pH* ≥ 5.1 while Ti–ATP complexes are formed at pH* ≤ 5.1. | ||
The 1H NMR assignments for free EHPG (rac and meso) ligands and TiIV–EHPG complexes have been established by 2D NMR techniques and their solid-state structures have been determined by X-ray crystallography.18 They are seven-coordinate with a hexadentate EHPG ligand and an additional water ligand. However, efforts to crystallise TiIV–ATP complexes were not successful and their structures cannot be established from the NMR data alone, although it is evident that both N7 and phosphate groups (β and γ) are involved in TiIV coordination.
At pH* 2.6, new 31P peaks emerged at δca. 0.70 and 0.81 (data not shown). These can be assigned to inorganic phosphate and AMP, respectively, indicating that the phospho-diester bonds of ATP are cleaved by reaction with titanocene dichloride.9 These results suggest that the affinity of Cp2TiCl2(aq) for EHPG or ATP is pH-dependent. At neutral pH, TiIV is more strongly bound to EHPG, while at acidic pH, it has a higher affinity for ATP. The cross-over point is at pH* ca. 5.1, which is comparable to the pH value inside the endosome (pH 5.0–5.5) where iron is released from transferrin.
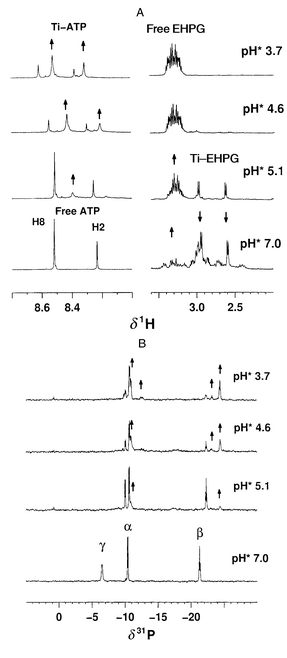
The Ti–EHPG (rac) complex, [Ti(ehpg)(H2O)] (1), is very stable between pH* 7 and 1,18 however, unexpectedly, in the presence of ATP, it becomes labile and TiIV transfers from the hexadentate ligand to ATP. Fig. 2 shows the time course of the reaction between 1 and ATP monitored by 1H and 31P NMR at 310 K and pH* 2.80. In the presence of 1 mol equiv. of ATP, 1H NMR peaks for 1 [δ 2.76 (d) and 3.12 (d) (methylene), δ 6.82 (d), 7.19 (t) and 7.50 (m) (phenyl ring)] decreased in intensity, while, simultaneously, new peaks characteristic of free EHPG [δ 3.40 (m) and 3.56 (m) (methylene), δ 7.05 (d), 7.10 (t), 7.35 (d), 7.46 (t) (phenyl ring)] appeared and increased in intensity with time. This indicates that TiIV dissociates from the EHPG ligand. At the same time, the H8, H2 peaks for free ATP decreased in intensity, while new H8 and H2 peaks appeared upfield and continued to increase in intensity. Also new 31P peaks emerged in the 31P NMR spectrum (δ 0.95, 0.82, −10.00, −10.47) and increased in intensity with time. These new peaks may be due to the species produced from the cleavage of ATP induced by TiIV (such as ADP, AMP and inorganic phosphate). Therefore TiIV is transferred from EHPG to ATP.
 | ||
| Fig. 2 Time course of the reaction between Ti(ehpg)(H2O) (1) and ATP (1∶1) at 310 K, pH* 2.80 followed by (A) 1H and (B) 31P{1H} NMR, showing the gradual dissociation of complex 1 accompanied by formation of Ti–ATP complexes, indicating intermolecular TiIV transfer. | ||
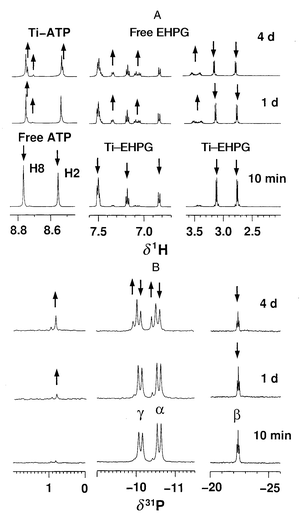
To investigate if intermolecular TiIV transfer can occur at physiologically relevant pH values, and the effect of ATP concentration, separate experiments were carried out in D2O at 310 K, using 1∶ATP mol ratios of 1∶1 or 1∶10, at pH* 2.8, 4.4 and 6.2. The simultaneous decrease in intensity of 1H NMR resonances for 1 and free ATP, and increase in intensity of resonances for free EHPG and Ti–ATP adducts, confirm that TiIV transfer does occur under these conditions. Fig. 3 shows the time course of TiIV transfer, as determined by integration of 1H NMR peaks. In the presence of 1 mol equiv. of ATP at pH* 2.8, 38% of the TiIV was transferred in 2 d; at pH* 4.4, 25% TiIV was transferred, while at pH* 6.2, little TiIV transfer was detected over a 2 d period. In the presence of 10 mol equiv. of ATP, TiIV transfer was more complete and even occurred at pH* 6.2. In 2 d, 83% TiIV was transferred at pH* 2.8, 41% at pH* 4.4, and 21% at pH* 6.2. A similar reaction was also carried out using 5′-GMP instead of ATP at pH* 2.9. TiIV transfer also occurred, though to a lower extent, in the presence of 5′-GMP (ca. 36% in the presence of 10 mol equiv. of 5′-GMP, data not shown).
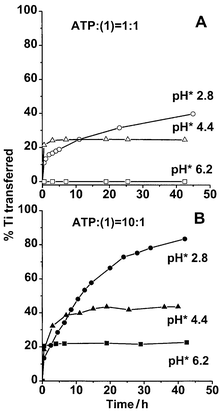 | ||
| Fig. 3 Time course of intermolecular TiIV transfer from complex 1 to ATP in the presence of (A) 1 mol equiv. ATP or (B) 10 mol equiv. ATP at different pH* values. The data show that both pH and ATP concentration affect the rate of TiIV transfer. | ||
Titanium transfer reactions to nucleotides mediated by EHPG, a chelator and amino acid derivative, may be relevant to the mechanism of action of titanium anticancer drugs. In terms of “HSAB” theory,20 TiIV is a “hard” Lewis acid and readily hydrolyses to form insoluble polymeric species at neutral pH values. Both titanocene dichloride and Budotitane undergo rapid and complete hydrolysis to form anticancer-inactive insoluble polymers at physiological pH values.1 However, biological experiments reveal that titanium is accumulated in the cellular nucleic-acid-rich regions (mainly the nucleus) after in vivo application of titanium drugs.7,8 Hence TiIV must be stabilised for transport by binding to biomolecules. Transferrin is a likely candidate for TiIV transport from blood plasma to cells.13,14 The current work shows that at neutral pH, Cp2TiCl2 has a higher affinity for the transferrin model ligand EHPG than for ATP while at pH* values below 5.1 the affinity for the nucleotide chelator ATP is higher. Intermolecular TiIV transfer from TiIV–EHPG complexes to nucleotides occurs at low pH or high ATP concentrations. TiIV transfer from human transferrin (Ti2–hTF) to ATP also occurs at low pH.14 Extra-cellular ATP levels are low, but intracellular concentrations of ATP are as high as 3–5 mM.21 Inside cells, ATP is a major metal macrochelator and intracellular iron carrier. It plays a major role in transport of FeIII to the nucleus, and the γ-phosphate of ATP is hydrolysed during FeIII transport.22 Therefore ATP could also facilitate the intracellular transport of TiIV and allow it to target polynucleotides which are condensed in the nucleus. DNA in the nucleus has a high negative charge and potentially a markedly lower pH value near its surface (up to 3 pH units lower than the bulk pH23). Cp2Ti–DNA adducts have been detected in vitro at pH 5.3,24 and Ti–DNA adducts in tumour cells treated with Cp2TiCl2.2
Metal anticancer agents are often electrophilic and can react with many biomolecules, such as amino acids, polyphosphates, proteins and nucleic acids. However, these biomolecules are located in different extracellular and intracellular compartments, and there are carrier molecules (e.g. proteins such as albumin and transferrin, or small molecules such ATP, citrate, and GSH) which communicate between them. The substrate binding properties (such as uptake and release) are finely controlled by natural gradients which exist in different tissues or cellular compartments (e.g. pH, ATP or ionic gradients). The gradients could also alter the relative affinity of drug molecules for different cellular components and facilitate drug binding to its target. “Hard” TiIV may be transported into the cell by transferrin and subsequently bind to DNA at both the negatively-charged phosphates on the backbone and base N-donors.6b,25 The high DNA concentration in the cell nucleus and potentially low pH close to the surface of DNA may favour DNA as a target for TiIV binding under these conditions. Further work is needed to establish this.
Acknowledgements
We thank the Committee of Vice-Chancellors and Principals for an ORS award and University of Edinburgh for a Scholarship (to M. G.) and BBSRC and EPSRC for support.References
- (a) P. Köpf-Maier and H. Köpf , in Metal Compounds in Cancer Therapy, ed. S. P. Fricker, Chapman & Hall, London, 1994, p. 109; Search PubMed; (b) B. K. Keppler , C. Friesen , H. Vongerichten and E. Vogel , in Metal Complexes in Cancer Chemotherapy, ed. B. K. Keppler, VCH, Weinheim, 1993, p. 297; Search PubMed; (c) P. Köpf-Maier and H. Köpf, Struct. Bonding (Berlin), 1988, 70, 103 Search PubMed.
- C. V. Christodoulou, A. G. Eliopoulos, L. S. Young, L. Hodgkins, D. R. Ferry and D. J. Kerr, Br. J. Cancer, 1998, 77, 2088 CAS.
- C. W. Schwietert and J. P. McCue, Coord. Chem. Rev., 1999, 184, 67 CrossRef CAS.
- I. C. Tornieporth-Oetting and P. S. White, Organometallics, 1995, 14, 1632 CrossRef CAS.
- (a) E. J. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467 CrossRef CAS; (b) J. Reedijk, Chem. Commun., 1996, 801 RSC; (c) J. Reedijk, Chem. Rev., 1999, 99, 2499 CrossRef CAS; (d) Z. Guo and P. J. Sadler , Angew. Chem., Int. Ed., 1999, 38, 4001 and refs. therein. Search PubMed.
- (a) L. Y. Kuo, A. H. Liu and T. J. Marks, Met. Ions Biol. Syst., 1996, 33, 53 Search PubMed; (b) P. Yang and M. Guo , Coord. Chem. Rev., 1999, 185–186, 189 and refs. therein. Search PubMed.
- P. Köpf-Maier and R. Martin, Virchows Arch. B, 1989, 57, 213 Search PubMed.
- P. Köpf-Maier, J. Struct. Biol., 1990, 105, 35 CrossRef CAS.
- M. Guo and P. J. Sadler , unpublished work. .
- M. M. Harding, G. Mokdsi, J. P. Mackay, M. Prodigalidad and S. W. Lucas, Inorg. Chem., 1998, 37, 2432 CrossRef CAS.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511 CrossRef CAS.
- J. H. Toney and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 947 CrossRef CAS.
- H. Sun, H. Li, R. Weir and P. J. Sadler, Angew. Chem., Int. Ed., 1998, 37, 1577 CrossRef CAS.
- M. Guo , H. Sun , P. J. Sadler , submitted..
- H. Sun , H. Li and P. J. Sadler , Chem. Rev., 1999, 99, 2817 Search PubMedand refs. therein..
- (a) V. L. Pecoraro, W. R. Harris, C. J. Carrano and K. N. Raymond, Biochemistry, 1981, 20, 7033 CrossRef CAS; (b) W. Lin, W. J. Welsh and W. R. Harris, Inorg. Chem., 1994, 33, 884 CrossRef CAS.
- H. Li, P. J. Sadler and H. Sun, Eur. J. Biochem., 1996, 242, 387 CrossRef CAS.
- M. Guo , H. Sun , S. Bihari , J. A. Parkinson , R. O. Gould , S. Parsons and P. J. Sadler , Inorg. Chem., 1999, Search PubMedin the press..
- (a) L. Y. Kuo, M. G. Kanatzidis and T. J. Marks, J. Am. Chem. Soc., 1987, 109, 7207 CrossRef CAS; (b) L. Y. Kuo, M. G. Kanatzidis, M. Sabat, A. L. Tipton and T. J. Marks, J. Am. Chem. Soc., 1991, 113, 9027 CrossRef CAS.
- R. G. Pearson , in Survey of Progress in Chemistry, ed. A. Scott, Academic Press, New York, 1969, ch. 1. Search PubMed.
- C. K. Mathews and K. E. van Holde , Biochemistry, The Benjamin/Cummings Pub. Co. Inc., Redwood City, USA, 1990, p. 410. .
- (a) P. Aisen, Adv. Exp. Med. Biol., 1994, 356, 31 Search PubMed; (b) S. A. Gurgueira and R. Meneghini, J. Biol. Chem., 1996, 271, 13616 CrossRef CAS.
- G. Lamm and G. R. Pack, Proc. Natl. Acad. Sci. USA, 1990, 87, 9033 CAS.
- M. L. McLaughlin, J. M. Cronan, Jr., T. R. Schaller and R. D. Sneller, J. Am. Chem. Soc., 1990, 112, 8949 CrossRef CAS.
- (a) M. Guo, P. Yang, B. Yang and Z. Zhang, Chin. Sci. Bull., 1996, 41, 1098 Search PubMed; (b) P. Yang and M. Guo, Met.-Based Drugs, 1998, 5, 41 Search PubMed.
Footnotes |
| † pH* is the pH meter reading in D2O solution. |
| ‡ EHPG represents the H4ehpg ligand without designation of the state of protonation. |
| This journal is © The Royal Society of Chemistry 2000 |