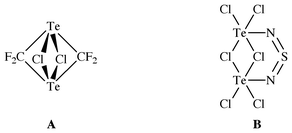Perfluoroalkyltellurocarbonyl fluorides, their cyclic dimers and perfluoroalkanetellurenyl iodides: preparation and reactivity![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) †
†
Received
(in Cambridge, UK)
17th September 1999
, Accepted 8th November 1999
First published on 14th January 2000
Abstract
With the synthesis of new perfluoroalkyltellurocarbonyls, their precursors and cyclic dimers additional valuable information about these classes of compounds has become available. They were prepared via pyrolysis of Me3SnTeCF(CF3)2 and the novel compounds Me3SnTeR (R = n-C3F7, n-C4F9). The monomer (CF3)2C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) Te could not be detected as it dimerises quantitatively to the corresponding 1,3-ditelluretane. It was possible to isolate R(F
Te could not be detected as it dimerises quantitatively to the corresponding 1,3-ditelluretane. It was possible to isolate R(F![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) )CTe (R = C2F5, n-C3F7) at −196 °C, but slightly above this temperature, these compounds cyclise to mixtures of cis/trans 1,3-ditelluretanes. The tellurocarbonyls undergo [4 + 2]-cycloaddition reactions with 2,3- dimethylbutadiene providing the corresponding tellurins. The reaction between RTeTeR and mercury yielded Hg(TeR)2 which have been converted with iodine in CH2Cl2 solution to RTeI. With AgCN the in situ prepared iodides form RTeCN (R = CF(CF3)2, n-C3F7, n-C4F9) in good yields. A new type of compound has been synthesized by chlorination of tetrafluoro-1,3-ditelluretane: 1,3-dichloro-2,2,4,4-tetrafluoro-1λ4,3λ4-ditellurabicyclo[1.1.0]butane. It is only stable below −20 °C and rearranges almost quantitatively at room temperature to ClF2CTeTeCF2Cl.
)CTe (R = C2F5, n-C3F7) at −196 °C, but slightly above this temperature, these compounds cyclise to mixtures of cis/trans 1,3-ditelluretanes. The tellurocarbonyls undergo [4 + 2]-cycloaddition reactions with 2,3- dimethylbutadiene providing the corresponding tellurins. The reaction between RTeTeR and mercury yielded Hg(TeR)2 which have been converted with iodine in CH2Cl2 solution to RTeI. With AgCN the in situ prepared iodides form RTeCN (R = CF(CF3)2, n-C3F7, n-C4F9) in good yields. A new type of compound has been synthesized by chlorination of tetrafluoro-1,3-ditelluretane: 1,3-dichloro-2,2,4,4-tetrafluoro-1λ4,3λ4-ditellurabicyclo[1.1.0]butane. It is only stable below −20 °C and rearranges almost quantitatively at room temperature to ClF2CTeTeCF2Cl.
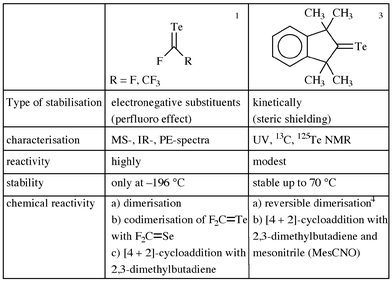
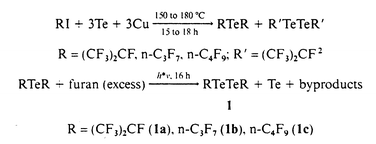
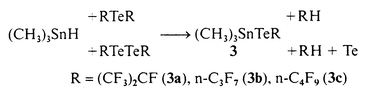
In 1991 the preparation and characterisation of the first compound with a CTe double bond was published. Meanwhile other examples were synthesized. Their stability is based either on the perfluoro effect or on bulky ligands. In Chart 1 the published tellurocarbonyls are listed with their known properties. The best synthesis for perfluorinated tellurocarbonyls is the pyrolysis of (CH3)3SnTeR (R = CF3,1 C2F5,2 made by reacting (CH3)3SnH with RfTexRf (x = 1, 2); methods which proved to be generally applicable. The required starting materials, such as RTexR (x = 1, R = (CF3)2CF, n-C3F7, n-C4F9, x = 2, R = (CF3)2CF,2 n-C3F7, n-C4F9) were made from RI and Te in the presence of copper as a catalyst.2 The ditellurides were also made by irradiating monotellurides dissolved in furan with UV-light as demonstrated in Scheme 1. The ditellurides 1a, 1b and 1c reacted
Metathesis of RTexR (x = 1, 2) with (CH3)3SnH provided (CH3)3SnTeR according to eqn. (2).
 |
| | Chart 1 | |
 |
| | Scheme 1 | |
| |
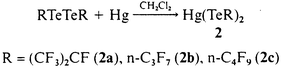
|
(1)
|
| |
|
(2)
|
The aim of this paper is: to prove that pyrolysis of (CH3)3SnTeR provides new highly reactive tellurocarbonyls, to study their chemistry and to synthesize perfluoroalkyltellurenyl reactants as versatile synthons.
Results and discussion
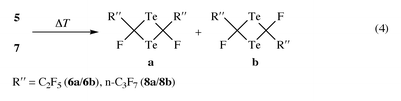
The pyrolysis of (CH3)3SnTeCF(CF3)2 at 500 °C and 10−3 Torr provided for the first time, compared with analogous reactions, not the monomer (CF3)2CTe, but its cyclic dimer. The very unstable intermediate (and there can be no doubt that it is formed primarily) condensed at −196 °C already in its dimeric form. With other perfluoroalkyl groups the corresponding monomers 5 and 7, formed according to eqn. (3), could be isolated and characterised by IR 5 and mass spectra 5, 7. On warming compounds 5 and 7 a few degrees above −196 °C a spontaneous change of colour from green to red was observed, yielding quantitative formation of the cis/trans-isomers of 6 and 8 [eqn. (4)].
| |
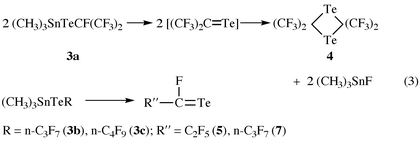
|
(3)
|
| |
|
(4)
|
With the aid of a special apparatus1,2 compound 5, generated by pyrolysis of 3a, was allowed to pass through an IR cell before being trapped at −196 °C. In this manner the IR spectrum of pure gaseous 5 was obtained. If the pyrolysis apparatus was coupled to the gas inlet of a mass spectrometer, the mass spectra of 5 and 7 could be registered. Although ν1 [νasm (CTe)] = 1240.0 cm−1 and ν4 [νasm (CF2)] = 1206.7, 1095.4 cm−1 were assigned to TeCF2,1 it was not possible to assign vibrations of strong to medium intensities in the area of 1257 to 1140 cm−1 for 5, because any assignment would have remained speculative and was therefore avoided. The mass spectra of compounds 5 and 7 showed M+ and the expected fragmentation pattern being different from those of the corresponding dimers 6a/6b and 8a/8b.
Chemical evidence for the formation of (CF3)2CTe, 5 and 7 comes from their dimerisation either immediately or by warming the product trap. The mixtures of cis/trans-isomers cannot be separated by physical methods, including preparative gas chromatography [eqn. (4)].
Crystal structure of compounds 4 and 4a
The compounds 4 and 4a are isostructural and both are isotypic to trans-[(F3C)(F)CSe]25 and [(F3C)(F)CTe]2.2
Views of the molecules are given in Fig. 1, selected bond lengths and angles in Table 1. Both molecules are centrosymmetric with molecular symmetry Ci and contain a planar C2Te2 ring. The transannular Te–Te distance is 3.246 Å for 4, and 3.1263 Å for 4a which is longer than a Te–Te single bond, e.g. 2.669 Å in (F3C)TeTe(CF3),6 but much less than the sum of the van der Waals radii (4.40 Å).7 Transannular Te–Te distances of these lengths are typical for molecules with C2Te2 rings: [(F3C)(F)CTe]2 3.271 Å, [F2CTe]2 3.385 Å.1a In the structure of 4a the molecules are present in the trans form. The C(1)–Cl bond of 1.77 Å is in the expected range. The structure determination did not reach very high levels of accuracy. Expecially the C(2)–F(2) bond with a length of 1.10 Å is unreasonably short. The peaks in the residual electron density map can be interpreted by positioning a second Cl atom near F(2) and a second CF3 group near Cl. The attempts to refine a disordered model with a superposed second molecule of [(F3C)CTe]2 in the ratio 80∶20 generated by a twofold rotation around the C(1)–C(1)′ axis resulted not in lowering the reliability factors but in some instability of the refinement. The marked effects of cis/trans disorder or dynamic exchange as observed in the crystals of [(F3C)(F)CTe]22 were not evident in the structure of 4a.
Table 1 Selected bond lengths (Å) and angles (°) for compounds 4 and 4a
| [(F3C)2CTe]24 |
[(F3C)(Cl)CTe]24a |
| Te–C(1) |
2.172(3) |
Te–C(1) |
2.16(1) |
| Te–C(1) |
2.171(3) |
Te–C(1)′ |
2.22(1) |
| C(1)–C(11) |
1.526(5) |
C(1)–Cl |
1.77(1) |
| C(1)–C(21) |
1.517(5) |
C(1)–C(2) |
1.58(3) |
| C(11)–F(11) |
1.325(5) |
C(2)–F(1) |
1.27(2) |
| C(11)–F(12) |
1.388(4) |
C(2)–F(2) |
1.10(2) |
| C(11)–F(13) |
1.314(5) |
C(2)–F(3) |
1.33(2) |
| C(21)–F(21) |
1.322(5) |
|
|
| C(21)–F(22) |
1.329(4) |
|
|
| C(21)–F(23) |
1.320(5) |
|
|
| |
|
|
|
| Te–C(1)–Te |
96.8(1) |
Te–C(1)–Te′ |
97.0 |
| C(1)–Te–C(1) |
83.2(1) |
C(1)–Te–C(1)′ |
83.0(5) |
| C(11)–C(1)–C(21) |
111.6(3) |
C(2)–C(1)–Cl |
109(1) |
| C(11)–C(1)–Te |
111.3(2) |
Te–C(1)–Cl |
112.5(7) |
| C(11)–C(1)–Te′ |
111.9(2) |
Te′–C(1)–Cl |
114.1(8) |
| C(21)–C(1)–Te |
113.1(2) |
C(2)–C(1)–Te |
112(1) |
| C(21)–C(1)–Te′ |
111.4(2) |
C(2)–C(1)–Te′ |
112(1) |
| C(1)–C(11)–F |
111.6(3)–112.8(3) |
C(1)–C(2)–F |
106(2)–111(2) |
| C(1)–C(21)–F |
111.9(3)–112.5(3) |
F–C(2)–F |
109(2)–110(2) |
| F–C(11)–F |
106.5(3)–107.0(4) |
|
|
| F–C(21)–F |
106.2(3)–106.9(3) |
|
|
![Molecular structures and atomic labelling scheme of [(F3C)2C Te]2 (4) (top) and [(F3C)(Cl)CTe]24a (bottom) with thermal ellipsoids shown at the 50% probability level.](/image/article/2000/DT/a907549f/a907549f-f1.gif) |
| | Fig. 1 Molecular structures and atomic labelling scheme of [(F3C)2C Te]2 (4) (top) and [(F3C)(Cl)CTe]24a (bottom) with thermal ellipsoids shown at the 50% probability level.
| |
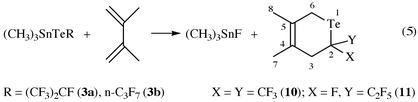
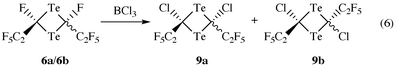
For further characterization of (CF3)2CTe as well as of compounds 5 and 7, [4 + 2]-cycloaddition reactions with 1,3-dimethylbutadiene have been carried out. The reaction took place in situ without isolation of the telluroketones following a procedure already employed for F2CTe1 and CF3(F)CTe.2 Heating compound 3a or 3b to 160 °C for 8 or 15 h dissolved in CHCl3 in the presence of a large excess of 2,3-dimethylbutadiene produces 3,6-dihydro-4,5-dimethyl-2,2-bis(trifluoromethyl)tellurin 10 or 2-fluoro-3,6-dihydro-4,5-dimethyl-2-(pentafluoroethyl)tellurin 11 according to eqn. (5). Compounds 10 and 11 are pale yellow oils of low volatility which are extremely sensitive to air. In solution they decompose at room temperature with separation of tellurium. Another general applicable reaction is the successful F/Cl exchange in 6a/6b with BCl3 occuring at the same rate to give a mixture of cis and trans forms of 9a, 9b [eqn. (6)]. So the mixture of 6a/6b could not be separated by use of the chemical reaction with BCl3.
| |
|
(5)
|
| |
|
(6)
|
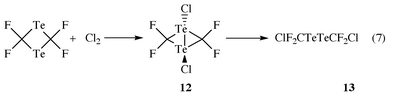
1,3-Dichloro-2,2,4,4-tetrafluoro-1λ4,3λ4-ditellurabicyclo[1.1.0]butane (12) and bis(chlorodifluoromethyl) ditelluride (13)
Chlorination of tetrafluoro-1,3-ditelluretane, dissolved in CH2Cl2 with excess Cl2 at −80 °C provides insoluble red-orange crystals of composition C2Cl2F4Te2. Spectroscopic investigations were applied for elucidating its structure. The NMR spectra, measured at −40 °C in acetonitrile solution, showed singlets at δF
−63.25, proving four equivalent fluorine atoms, δC 84.09 (m) and a quintet at δTe 2009.00 showing equivalent carbon and tellurium atoms. The mass spectrum evidenced M+ and the expected fragmentation pattern containing TeCl+, but no TeCl2+ fragments. These results are in good agreement with a planar C2Te2-ring frame having the two Cl-atoms at each Te-atom in a trans-position. The oxidative chlorination leads also to a Te–Te bond yielding bicyclic 12. Conclusive evidence is also obtained by its instability. On the basis of NMR-arguments, structure A cannot be ruled out, as such Te⋯Cl⋯Te bonding had been observed for compound B.8,9 At room temperature orange-red 12, dissolved in CH3CN, changed colour to dark brown. Fractional condensation at 20 °C in vacuo gave an orange liquid condensate at −50 °C. Spectroscopic investigations and a quantitative chlorine analysis evidenced the formation of ClF2CTeTeCF2Cl 13 as shown in eqn. (7).
| |
|
(7)
|
Oxidation of the two tellurium atoms in [F2CTe]2 by chlorine are accompanied by a shift of δTe 313.0 to lower field compared with δTe, of the starting material. This effect was also observed in other similar chlorination processes. When (CF3)2Te was chlorinated to (CF3)2TeCl2, δTe was shifted 254 ppm to lower field from δTe 1368 to δTe 1114.10
An interesting class of compounds are the perfluoroalkyltellurenyl iodides 14. Although only stable in solution, they are versatile reactants, e.g. metathetical reactions, and were made from compounds 2a, 2b or 2c and I2, dissolved in CH2Cl2, reacting with for example AgCN to give the corresponding cyano derivatives 15 according to eqn. (8).
| |
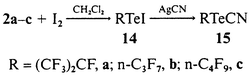
|
(8)
|
Experimental
All reactions were carried out in a standard vacuum system with Teflon-stemmed Young valves; solvents were dried according to published procedures. Deuteriated solvents were dried and transferred from activated 400 pm sieves. Microanalyses were performed on a 1106 Carlo-Erba Elemental analyser. The NMR spectra were recorded for CDCl3 solutions unless noted otherwise, using a Bruker WM 250 PFT spectrometer [standards: CDCl3 (13C), SiMe4 (1H), CFCl3 (19F), TeMe2 (125Te) and SnMe4 (119Sn)], infrared spectra on a Bruker FT-IR IFS 66, Raman spectra using a Raman attachment (FRA 106) to the Bruker FT-IR IFS 66 and mass spectra were recorded on a MAT CH7 spectrometer, using the direct-inlet method with 70 eV ionisation radiation. Compound 4a was synthesized according to the literature2 method.
Crystal structure determination of compounds 4 and 4a
Crystals of 4 and 4a were selected under paraffin and placed in 0.3 mm glass capillaries which were flame sealed. Data collections were performed with a STOE AED 2 four-circle diffractometer using Mo-Kα radiation. Numerical absorption corrections were applied to both data sets. The crystal shapes were optimised by minimisation of Rint (HABITUS,114: transmission factors 0.6106–0.6878, 4a: 0.4133–0.6030). The structures were solved by direct methods (SHELXS-86)12 and refined by full matrix least squares on F2 (SHELXL-93)13 with anisotropic displacement parameters for all atoms. Crystallographic data are given in Table 2.
Table 2 Experimental details of the crystallographic studies
| |
[(CF3)2CTe]2 (4) |
[(CF3)(Cl)CTe]2 (4a) |
| Chemical formula |
C6F12Te2 |
C4Cl2F6Te2 |
| Formula weight |
555.24 |
488.14 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
|
μ(Mo-Kα)/cm−1 |
48.8 |
59.9 |
|
a/Å |
7.6033(6) |
7.771(7) |
|
b/Å |
7.2595(6) |
6.749(2) |
|
c/Å |
11.5378(8) |
10.685(2) |
|
β/° |
105.715(7) |
105.30(4) |
|
V/Å3 |
613.0 |
540.5 |
|
Z
|
2 |
2 |
|
T/K |
295 |
295 |
| No. data collected |
7372 |
6493 |
| No. unique data |
1789 |
1566 |
|
R
int
|
0.027 |
0.061 |
| Final R(F2) for all data |
0.056 |
0.250 |
| Final R(|F|) for Fo > 4σ(Fo) |
0.025 |
0.125 |
CCDC reference number 186/1729.
See http://www.rsc.org/suppdata/dt/a9/a907549f/ for crystallographic files in .cif format.
Bis(perfluoroisopropyl)- (1a), bis(perfluoro-n-propyl)- (1b) and bis(perfluoro-n-butyl)-ditelluride (1c)
In a quartz Carius tube (200 cm3) equipped with a Teflon-stemmed Young valve and a magnetic stirring bar 15 ml of furan and (n-C3F7)2Te (5.21 g, 11.20 mmol) or (n-C4F9)2Te (4.30 g, 7.60 mmol) respectively were condensed in vacuo at −196 °C and degassed. The mixtures were irradiated at 20 °C (16 h) with a UV-lamp (type Heraeus TQ 150, 250 nm). Separation of the products was accomplished by fractional condensation using U-shaped traps kept at 20, −40 or −5 and −196 °C. Pure 1b (2.42 g, 73%) condensed at −40 °C or 1c (1.19 g, 45%) at −5 °C respectively as yellow-red liquids. 1b: (found: C, 12.3. C4F14Te2 requires C, 12.1%). 1c: (found: C, 14.2. C8F18Te2 requires C, 13.9%). 1a: physical data are identical with published ones.2 See SUP data for IR, 13C, 19F, 125Te NMR and MS data for 1b and 1c.
Bis(perfluoroalkanetellurenyl)mercury (2)
These compounds were synthesized in a glass apparatus fitted with two 50 ml Carius tubes closed with Teflon valves and a medium sintered glass frit. One tube contained a magnetic stirring bar and a solution of the ditelluride in 10 ml CH2Cl2. A threefold excess of mercury was added under argon and stirred at 20 °C for 1 h. The yellow solution was filtered into the other Carius tube and evaporated in vacuo to dryness yielding yellow Hg(TeRf)2. The compounds Hg(TeRf)2, Rf = (CF3)2CF (2a), n-C3F7 (2b) and n-C4F9 (2c) are made from (2.10 g, 3.54 mmol) (CF3)2CFTeTeCF(CF3)2, (2.42 g, 4.08 mmol) 1a and (1.26 g, 1.82 mmol) 1b yielding 2a (2.54 g, 90%), 2b (2.87 g, 89%) and 2c (1.47 g, 90%) respectively. 2a: (found: C, 8.6. C6F14HgTe2 requires 9.1%). 2b: (found: C, 8.7. C6F14HgTe2 requires 9.1%). 2c: (found: C, 10.3. C8F18HgTe2 requires 10.8%). See SUP data for IR, 13C, 19F, 125Te NMR and MS data for 2a–c.
Perfluoroalkyltrimethylstannyltelluride (3)
In a Carius tube (50 ml) equipped with Teflon-stemmed Young valves and magnetic stirring bar (n-C3F7)2Te (1.80 g, 3.87 mmol) or (n-C4F9)2Te (3.50 g, 6.19 mmol) respectively was dissolved in diethyl ether (10 cm3). The yellow solution was cooled to −196 °C and Me3SnH (0.64 g, 3.87 mmol or 1.02 g, 6.19 mmol respectively) was condensed in. The mixture was warmed under the exclusion of light to −15 or 0 °C with stirring for 1 or 2 h respectively, changing from bright yellow to pale yellow. After trap to trap condensation in vacuo (−40 to −196 or −15 to −196 °C respectively) Me3SnTe(n-C3F7) (3b, 1.16 g, 65%) or Me3SnTe(n-C4F9) (3c, 1.64 g, 52%) were obtained as pale yellow, light sensitive liquids. 3b: (found: C, 15.4; H, 2.1. C6H9F7SnTe requires C, 15.7; H, 2.0%). 3c: (found: C, 16.6; H, 1.8. C7H9F9SnTe requires C, 16.5; H, 1.8%).
From Te2(Rf)2..
Compound 1b (0.83 g, 1.40 mmol) or compound 1c (1.30 g, 1.88 mmol) was treated with Me3SnH (0.23 g, 1.40 mmol or 0.31 g, 1.88 mmol). After purification 3b (0.48 g, 75%) or 3c (0.39 g, 41%) was obtained. See SUP data for IR, 1H, 13C, 19F, 119Sn, 125Te NMR and MS data for 3b and 3c.
A sample of Me3SnTeCF(CF3)2 (2.15 g, 4.67 mmol) was passed at 10−3 Torr and 550 °C through a quartz-glass pyrolysis tube (20 cm, diameter 1 cm). This was connected to two U-tubes, the first cooled to −30 °C and the second to −196 °C. The pyrolysis reaction took 1.5 h. In the U-tube at −30 °C some Me3SnF and unchanged Me3SnTeCF(CF3)2 were retained. The main amount of Me3SnF sublimed to the cooler zone of the quartz tube. In the following trap 4 was condensed as a dark red solid which was purified by sublimation (0.73 g, 56%). It is stable at 20 °C, not air sensitive and soluble in common organic solvents. The formation of the monomeric intermediate (CF3)2CTe could not be detected. Mp 113 °C (Found: C, 13.1. C6F12Te2 requires C, 13.0%). See SUP data for IR, Raman, 13C, 19F, 125Te NMR and MS data for 4.
Pentafluoroethyltellurocarbonyl fluoride (5)
This compound was prepared as described for 4 by pyrolysing 3a (1.87 g, 4.06 mmol) at 10−3 Torr and 500 °C. In the U-tube, cooled to −40 °C, unchanged 3a was retained. In the following trap 5 was condensed as a green solid material. (In order to avoid dimerisation the liquid nitrogen surface should be kept higher than the condensation zone.) Experimental details for the characterisation of perfluorinated telluroketons are provided elsewhere.1,2 See SUP data for IR and MS data for compound 5.
cis/trans-2,4-Difluoro-2,4-bis(pentafluoroethyl)-1,3-ditelluretane (6a/6b)
A sample of compound 5, obtained in a trap cooled to −196 °C was warmed by removing the liquid nitrogen Dewar. A spontaneous change in the deposit from green to red was observed, yielding quantitative formation of 6. Compound 6 separated from trace impurities via fractional condensation in vacuo with the sample at ambient temperature. Fractions were collected at −40 and −196 °C. The product 6a/6b (0.72 g, 64% based on 3a) was found to condense at −40 °C as an air sensitive violet liquid, which is stable at 20 °C under argon for a few days. The cis and trans isomers could not be separated either by physical or by chemical methods. 6a/6b: (found: C, 13.0. C6F12Te2 requires C, 13.0%). See SUP data for IR, Raman, 13C, 19F, 125Te NMR and MS data for 6a/6b.
Heptafluoropropyltellurocarbonyl fluoride (7)
Compound 7 was prepared as described for 5 from 3b (1.30 g, 2.55 mmol). After 1.5 to 2 h the monomer 7 condensed at −196 °C as a green glassy compound which was characterised by its mass spectrum (see SUP data). Unchanged 3b was retained at −10 °C.
cis/trans-2,4-Difluoro-2,4-bis(heptafluoropropyl)-1,3-ditelluretane (8a/8b)
Isomers 8a/8b were made as described for 6a/6b and purified by fractional condensation in vacuo at 20 °C. Compound 8a/8b (0.41 g, 49% based on 3b) condensed at −40 °C as a violet solid. At 22 °C the deep violet liquid was stable for a few days under exclusion of air (Found: C, 14.5. C8F16Te2 requires C, 14.7%). See SUP data for IR, Raman, 13C, 19F, 125Te NMR and MS data for 8a/8b.
cis/trans-2,4-Dichloro-2,4-bis(pentafluoroethyl)-1,3-ditelluretane (9a/9b)
A sample of compound 6a/6b (0.15 g, 0.27 mmol) was deposited under an atmosphere of argon in a Carius tube (100 ml) and cooled to −196 °C. All argon was removed in vacuo and BCl3 (3 cm3) was added to the reactor. The mixture was initally warmed to −40 °C and thereafter the temperature was raised slowly to 22 °C over a period of 12 h. At −40 °C 6a/6b began to dissolve in BCl3 to give a deep violet solution. At room temperature the mixture turned blue. Compound 9a/9b was isolated by fractionation in vacuo. The product was collected in the trap cooled to −20 °C as violet crystals (0.13 g, 82%). It was not possible to separate the cis and trans isomers of 9, so all analyses were obtained on an isomeric mixture of 9a and 9b. See SUP data for 19F and MS data for 9a/9b.
3,6-Dihydro-4,5-dimethyl-2,2-bis(trifluoromethyl)tellurin (10)
Compound 10 was obtained by the direct reaction of (CH3)3SnTeCF(CF3)2 (2.00 g, 4.34 mmol) with 2,3-dimethylbutadiene (1.09 g, 13.3 mmol) dissolved in CHCl3 (5 ml) in a sealed glass ampoule (30 cm3). The mixture was heated to 160 °C for 8 h. Compound 10 was isolated by fractional condensation in vacuo and collected at −30 °C as a bright yellow, light and moisture sensitive liquid (1.05 g, 67%) (Found: C, 30.2; H, 2.7. C9H10F6Te requires C, 30.0; H, 2.8%). See SUP data for IR, 1H, 13C, 19F, 125Te NMR and MS data for 10.
2-Fluoro-3,6-dihydro-4,5-dimethyl-2-pentafluoroethyltellurin (11)
This compound was prepared as described for 10 by reacting excess 2,3-dimethylbutadiene (0.36 g, 4.42 mmol) with 3a (1.06 g, 2.30 mmol) in CHCl3 (5 ml) at 160 °C for 15 h. Compound 11 (0.62 g, 75%) was isolated by fractional condensation in vacuo at −30 °C as a bright yellow, moisture sensitive liquid, which slowly decomposed at 20 °C in solution depositing tellurium (Found: C, 29.8; H, 3.1. C9H10F6Te requires C, 30.1; H, 2.8%). See SUP data for IR, 1H, 13C, 19F, 125Te NMR and MS data for 11.
1,3-Dichloro-2,2,4,4-tetrafluoro-1λ4,3λ4-ditellurabicyclo[1.1.0]butane (12)
In a Carius tube (50 ml) equipped with a Teflon-stemmed Young valve and a magnetic stirring bar 2,2,4,4-tetrafluoro-1,3-ditelluretane (43.4 mg, 0.122 mmol) was deposited and dissolved in CH2Cl2 (1 cm3). The dark blue solution was cooled to −196 °C and evacuated. Thereafter Cl2 (26 mg, 0.733 mmol) was condensed in. The mixture was warmed to −80 °C and stirred for 1 h. Solvent and excess Cl2 were removed at −50 °C in vacuo providing temperature sensitive red-orange 12 (51.0 mg, 98%) soluble in CH3CN. Decomposition temperature −15 °C. See SUP data for IR, 13C, 19F, 125Te NMR and MS data for 12.
Bis(chlorodifluoromethyl)ditelluride (13)
A sample of compound 12, obtained in a Carius tube cooled to −50 °C, was warmed to room temperature and kept for 1 h. A colour change from red-orange to dark brown was observed. Compound 13 was purified by fractional condensation at ambient temperature. Fractions were collected at −50 and −196 °C. The product (47.0 mg, 90%) was found to condense at −50 °C as a bright yellow liquid changing to orange on warming to 20 °C. It is soluble in common solvents and decomposed slowly depositing tellurium. Mp −56 °C (Found: Cl, 16.0. C2Cl2F4Te2 requires Cl, 16.6%). See SUP data for IR, 13C, 19F, 125Te NMR and MS data for 13.
Perfluoroalkanetellurenyl iodides RfTeI [Rf = (CF3)2CF, 14a; n-C3F7, 14b; n-C4F9, 14c]
General procedure: in a carefully dried Carius tube (100 cm3) equipped with a Teflon-stemmed valve and magnetic stirring bar solutions of [(CF3)2CF]2Te2, 1a, 1b or 1c dissolved in CHCl3 (10 cm3) were deposited together with equivalent amounts of I2 under an argon atmosphere. The solution was cooled to −196 °C and the argon removed in vacuo. With continous stirring, the reaction mixture was annealed to room temperature; in the process it was observed to turn blue. The 19F NMR spectrum of the solution indicated that 14a, b or c had been formed quantitatively. The solutions were found to be stable at −25 °C over long periods. 14a: [(CF3)2CF]2Te2 (1.19 g, 2.01 mmol) and I2 (0.51 g, 2.01 mmol). 14b: (n-C3F7)2Te2 (1.28 g, 2.16 mmol) and I2 (0.55 g, 2.16 mmol). 14c: (n-C4F9)2Te2 (1.95 g, 2.81 mmol) and I2 (0.71 g, 2.81 mmol). See SUP data for 13C, 19F, 125Te NMR and MS data for 14a–c.
Cyano(perfluoroalkyl)tellurium [(CF3)2CFTeCN, 15a; n-C3F7TeCN, 15b; n-C4F9TeCN, 15c]
General procedure: to samples of compounds 14a, b or c obtained at −196 °C under argon equivalent amounts of AgCN were added. Thereafter argon was removed in vacuo and the mixtures were warmed to 22 °C with stirring. Yellow AgI desposited and the solution became green. The compounds were isolated by fractional condensation in vacuo with the samples at ambient temperature. Fractions were collected at −40 and −196 °C. The products were found to condense at −40 °C as air stable, slightly yellow coloured compunds soluble in common solvents. 15a: (CF3)CFTeI (0.63 g, 1.96 mmol) and AgCN (0.26 g, 1.96 mmol) provided 15a (0.51 g, 81%) (Found: C, 15.3; N, 4.5. C4F7NTe requires C, 14.9; N, 4.3%). 15b: n-C3F7TeI (1.83 g, 4.32 mmol) and AgCN (0.58 g, 4.32 mmol) gave n-C3F7TeCN (1.28 g, 92%), mp 89 °C (Found: C, 14.4; N, 3.9. C4F7NTe requires C, 14.9; N, 4.3%). 15c: n-C4F9TeI (2.67 g, 5.62 mmol) and AgCN (0.75 g, 5.63 mmol) provided n-C4F9TeCN (1.86 g, 89%); mp 98 °C (Found: C, 16.0; N, 3.7. C5F9NTe requires C, 16.1; N, 3.8%). See SUP data for IR, 13C, 19F, 125Te NMR and MS data for 15a–c.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie for financial support.
References
-
(a) R. Boese, A. Haas and C. Limberg, J. Chem. Soc., Dalton Trans., 1993, 2547 RSCsee also
(b) R. Boese, A. Haas and C. Limberg, J. Chem. Soc., Chem. Commun., 1991, 1379 Search PubMed;
(c) A. Haas and C. Limberg, Chimia, 1992, 46, 78 Search PubMed.
- J. Beck, A. Haas, W. Herrendorf and H. Heuduk, J. Chem. Soc., Dalton Trans., 1996, 4463 RSC.
- M. Minoura, T. Kawashima and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 7019 CrossRef CAS.
- M. Minoura, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1997, 38, 2501 CrossRef CAS.
- R. Boese, A. Haas and M. Spehr, Chem. Ber., 1991, 124, 51 CAS.
- W. Dukat, F. Gall, C. Meyer, D. Mootz, D. Naumann, G. Nowicki and K. Schulz, Z. Kristallogr., 1996, 622, 617 Search PubMed.
-
L. Pauling
, The Nature of the Chemical Bond, Cornell University Press, Ithaca, 1960.
Search PubMed.
- A. Haas, J. Kasperowski and M. Pryka, Chem. Ber., 1992, 125, 1537 CAS.
- A. Haas and M. Pryka, Chem. Ber., 1995, 128, 11 CAS.
- W. Gombler, Z. Naturforsch., Teil B, 1981, 36, 535 Search PubMed.
-
W. Herrendorf
and H. Bärninghausen
, HABITUS, Program for the optimisation of the crystal shape for numerical absorption correction, Universities of Karlsruhe and Giessen, 1993/1998.
Search PubMed.
-
G. M. Sheldrick
, SHELXS-86, Program for crystal structure solution, University of Göttingen, 1986.
Search PubMed.
-
G. M. Sheldrick
, SHELXL-93, Program for crystal structure refinement, University of Göttingen, 1993.
Search PubMed.
Footnote |
| † Supplementary data available: rotatable 3-D crystal structure diagram in CHIME format. See http://www.rsc.org/suppdata/dt/a9/a907390f/Also available: NMR, IR and mass spectroscopic data for compounds 1b–15c. See http://www.rsc.org/suppdata/dt/a9/a907549f/. See Instructions for Authors, 2000, Issue 1 (http://www.rsc.org/dalton). |
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.