Ruthenium(III)–aminopolycarboxylato complexes active for the reduction of the N–N bond of hydrazine and phenylhydrazine in aqueous acidic media
Received
(in Cambridge, UK)
4th October 1999
, Accepted 29th October 1999
First published on 14th January 2000
Abstract
Interactions of hydrazines N2H4X+ (X = H or Ph) with tri-, tetra- and penta-chelated ruthenium(III)–aminopolycarboxylic acid complexes giving the respective monomeric hydrazinium (RuIII–N2H4X+) adducts have been investigated by potentiometry, spectrophotometry and voltammetry in aqueous acidic solution at 25 °C. The deprotonation and metal hydrolysis constants of the complexes and their N2H4X+ adducts in 0.1 M Na2SO4 solution were determined. At pH 2.8, the complexes exhibited a quasi-reversible one-electron reduction wave of RuIII → RuII in sampled dc in the potential range between −0.16 and −0.37 V vs. SCE, while their hydrazinium adducts obtained in situ by adding an excess of N2H4X+ showed an additional two-electron reduction wave assigned to RuIII–N2H4X+ → RuI–N2H4X+ in the potential range of −0.02 to −0.35 V vs. SCE. The species RuI–N2H4X+ on successive decomposition and hydrolysis give one mole of each of NH3, NH2X and ruthenium(III) species. Further, the RuIII–N2H4X+ complexes have been used as electro-catalysts for the reduction of N2H4X+ to NH3 and NH2X at a mercury pool cathode in acidic solutions of pH 1.9 and 2.8. The quantity of ammonia produced in all cases is linear with time. The E1/2 of RuIII–N2H4X+ → RuI–N2H4X+ and the turnover number are correlated with the sigma basicity (ΣpKa) of the aminopolycarboxylic acids and the results are discussed in terms of the hydrolytic tendency of the metal, the number of co-ordinating groups and the steric repulsion caused by the increase in size of the aminopolycarboxylic acid.
Introduction
Studies on the reactivity of hydrazine at the potential co-ordination site of an active metal centre in the absence and the presence of reducing agents have immense value, since the reduction of dinitrogen to ammonia catalysed by transition metals present in microorganisms![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 1–7 or simple metal complexes or clusters has long been proposed to proceed via metal bound hydrazine. In the recent past intensive research has been carried out to evolve a simple mechanism for dinitrogen fixation. In this respect, numerous transition metals, cofactors and model compounds containing hydrazine were synthesized and characterized.8–10 Structural analyses of a few of these complexes have also been made.8,11 Meanwhile, reduction studies were also performed on hydrazine or its derivatives to ammonia and/or amine in both aqueous and non-aqueous solutions either in the absence or in the presence of external proton and electron sources or reducing agents.12–14 Although, fairly good turnovers of ammonia have reportedly been obtained in organic solvents, most of these reactions did not meet much success in aqueous solutions. However, Schrauzer et al.15 reported the reduction of hydrazine to ammonia using K2[Mo(O)(H2O)(CN)4] as a catalyst and NaBH4 as reducing agent with a turnover rate of 4.2 in aqueous solutions. Later catalytic conversion of hydrazine into ammonia was obtained by [Mo2Fe6S8L9]3− or [Fe4S4L4]2− (L = SPh or SCH2CH2OH) with a moderate turnover number.16 In another report,17 [WCp*Me3(η2-NH2NH2)][O3SCF3] prepared from [WCp*Me3(O3SCF3)] and N2H4 was shown to undergo reduction by Na–Hg to give NH3. Recently, the co-ordinatively unsaturated diruthenium complex [Cp*Ru(μ-SPri)2RuCp*] has been reported to catalyse the reduction of hydrazine more effectively.18 More sophisticated systems [MoFe3S4Cl3(Cl4C6O2)(MeCN)]2− and [MoFe3S4Cl3(citr)]3− (citr = citrate) were also modelled and used to catalyse the reduction of N2H4 to NH3 with good turnovers by CoCp2 and 2,6-dimethylpyridine hydrochloride.13,19 Moreover, the N2H4-bridged double cubane [{MoFe3S4Cl3(Cl4C6O2)}2(μ-NH2NH2)] was isolated and its catalytic activity studied.20 However, the parallel evolution of hydrogen gas which reduces the yield of NH3 was reported as the major limiting step in almost all the above systems.
1–7 or simple metal complexes or clusters has long been proposed to proceed via metal bound hydrazine. In the recent past intensive research has been carried out to evolve a simple mechanism for dinitrogen fixation. In this respect, numerous transition metals, cofactors and model compounds containing hydrazine were synthesized and characterized.8–10 Structural analyses of a few of these complexes have also been made.8,11 Meanwhile, reduction studies were also performed on hydrazine or its derivatives to ammonia and/or amine in both aqueous and non-aqueous solutions either in the absence or in the presence of external proton and electron sources or reducing agents.12–14 Although, fairly good turnovers of ammonia have reportedly been obtained in organic solvents, most of these reactions did not meet much success in aqueous solutions. However, Schrauzer et al.15 reported the reduction of hydrazine to ammonia using K2[Mo(O)(H2O)(CN)4] as a catalyst and NaBH4 as reducing agent with a turnover rate of 4.2 in aqueous solutions. Later catalytic conversion of hydrazine into ammonia was obtained by [Mo2Fe6S8L9]3− or [Fe4S4L4]2− (L = SPh or SCH2CH2OH) with a moderate turnover number.16 In another report,17 [WCp*Me3(η2-NH2NH2)][O3SCF3] prepared from [WCp*Me3(O3SCF3)] and N2H4 was shown to undergo reduction by Na–Hg to give NH3. Recently, the co-ordinatively unsaturated diruthenium complex [Cp*Ru(μ-SPri)2RuCp*] has been reported to catalyse the reduction of hydrazine more effectively.18 More sophisticated systems [MoFe3S4Cl3(Cl4C6O2)(MeCN)]2− and [MoFe3S4Cl3(citr)]3− (citr = citrate) were also modelled and used to catalyse the reduction of N2H4 to NH3 with good turnovers by CoCp2 and 2,6-dimethylpyridine hydrochloride.13,19 Moreover, the N2H4-bridged double cubane [{MoFe3S4Cl3(Cl4C6O2)}2(μ-NH2NH2)] was isolated and its catalytic activity studied.20 However, the parallel evolution of hydrogen gas which reduces the yield of NH3 was reported as the major limiting step in almost all the above systems.
Recently, we have reported the electrochemical reduction of hydrazine and its phenyl derivative to NH3 and/or NH2X at high turnover rates and high coulombic efficiency21–24 without the liberation of hydrogen gas using terminally co-ordinated RuIII–edta–N2H4X+ complexes as electro-catalysts in aqueous acidic solutions. In continuation, herein we describe the interaction of N2H4X+ (X = H or Ph) with tri-, tetra- and penta-chelated aminopolycarboxylates of ruthenium(III), viz. K2[Ru(imda)Cl3]·2H2O 1, K2[Ru(himda)Cl2]·H2O 2, K2[Ru(nta)Cl2]·H2O 3, K[Ru(hedta)Cl] 4, K[Ru(Hedta)Cl]·2H2O 5, K[Ru(Hpdta)Cl]·2H2O 6, K[Ru(Hcdta)Cl]·H2O 7 and K[Ru(H2dtpa)Cl] 8 by potentiometry, spectrophotometry and voltammetry in aqueous solution. The function of the resulting hydrazinium complexes as catalysts for the reduction of hydrazine and its phenyl derivative electrochemically in acidic solution is elucidated. The effects of the sigma basicity (ΣpKa) of the aminopolycarboxylic acid in complexes 1–8 on the half-wave potential (E1/2) of the two-electron reduction wave of RuIII–N2H5+ complexes and the turnover number of NH3 produced in the electrolytic reduction of N2H4X+ have been investigated.
Experimental
Ruthenium trichloride (RuCl3·xH2O) from Arora Matthey India, iminodiacetic acid (H2imda), N-(2-hydroxyethyl)iminodiacetic acid (H2himda), nitrilotriacetic acid (H3nta), N-carboxymethyl-N′-(2-hydroxyethyl)ethylenediiminodiacetic acid (H3hedta), propylenedinitrilotetraacetic acid (H4pdta), (±)-trans-cyclohexane-1,2-diyldinitrilotetraacetic acid (H4cdta), and carboxymethyliminobis(ethylenenitrilo)tetraacetic acid (H5dtpa) from Aldrich Chemical Company Inc. USA were purchased. Ethylenedinitrilotetraacetate disodium salt (Na2H2edta) from Polypharm India, hydrazine sulfate (N2H5HSO4) from BDH Chemicals, India and phenylhydrazine hydrochloride (N2H4PhCl) from Allied Chemicals, USA. The zero grade Ar gas from IOLAR & Co. was used after purification with vanadium sulfate and alkaline pyrogallol solutions. All other reagents used were of AR grade. A mixture of 0.2 M CH3COONa and 18 N H2SO4 solutions at a desired pH between 1 and 5 was used as supporting electrolyte in all voltammetric studies. Stock solutions of N2H5HSO4 (0.1 M) were prepared and standardized with potassium iodate.21 A freshly prepared solution of 0.1 M N2H4PhCl was used to prepare the experimental solutions.
Preparations
K2[RuCl5(OH2)]..
The compound was prepared by the method reported elsewhere,25 and used as the precursor to synthesize three- to five-co-ordinated ruthenium(III)–aminopolycarboxylic acid complexes 1–8 according to the following methods.
K2[Ru(imda)Cl3]·2H2O 1..
To a hot solution of 10 mL K2[RuCl5(OH2)] (0.375 g, 1 mmol) in 1 mM HClO4, H2imda (0.136 g, 1.02 mmol) dissolved in 10 mL of 1 mM HClO4 was added slowly with constant stirring. The resultant mixture was refluxed (two hours) till the reddish brown colour changed to pale yellow. Later, the volume of the solution was reduced (5 mL) in vacuo on a Rotovapour. It was then precipitated with cold absolute alcohol, filtered off, washed with 9∶1 acetone–water till free of chloride and dried under vacuum. Calc. for C4H9Cl3K2NO6Ru: C, 10.46; H, 1.92; N, 3.07. Found: C, 10.60; H, 2.01; N, 3.09%. IR (KBr): 1635 (COO− asym); 1365 (COO− sym); 1210 (CO); 840 (OCO def); 535 cm−1 (M–N). UV-vis, λ/nm (ε/M−1 cm−1): 240 (2270); 285 (1400) and 365 (445).
K2[Ru(himda)Cl2]·H2O 2..
The compound 2 was prepared from K2[RuCl5(OH2)] (0.375 g, 1 mmol) and H2himda (0.180 g, 1.02 mmol) by the procedure described for 1. Calc. for C6H10Cl2K2NO6Ru: C, 16.04; H, 2.32; N, 3.09. Found: 16.29; H, 2.28; N, 3.17%. IR (KBr): 1640 (COO− asym); 1365 (COO− sym); 1235 (CO); 825 (OCO def); 535 cm−1 (M–N). UV-vis, λ/nm (ε/M−1 cm−1): 245 (2595); 285 (2360) and 360 (640).
K2[Ru(nta)Cl2]·H2O 3..
This compound was prepared from K2[RuCl5(OH2)] (0.375 g, 1 mmol) and H3nta (0.195 g, 1.02 mmol) in the same manner as described above for 1. Calc. for C6H8Cl2K2NO7Ru: C, 15.85; H, 1.68; N, 3.13. Found: C, 15.79; H, 1.76; N, 3.07%. IR (KBr): 1635 (COO− asym); 1358 (COO− sym); 1230 (CO); 850 (OCO def); 525 cm−1 (M–N). UV-vis, λ/nm (ε/M−1 cm−1): 285 (2420) and 360 (735).
K[Ru(HmL)Cl]·xH2O {m = 0, x = 0 for L = hedta 4; m = 1, x = 2 for edta 5; m = 1, x = 2 for pdta 6}..
These compounds were prepared from K2[RuCl5(OH2)] and the respective aminopolycarboxylic acid according to the procedures reported earlier26–28 and subsequently characterized by physico-chemical methods.
K[Ru(Hcdta)Cl]·H2O 7..
This complex was obtained by treating K2[RuCl5(OH2)] (0.375 g, 1.0 mmol) in 10 mL 1 mM HClO4 with Na2H2cdta (0.400 g, 1.02 mmol) in 20 mL 1 mM HClO4 under a nitrogen atmosphere, initially for 30 min with stirring at room temperature followed by refluxing for about two hours. The greenish yellow solution was evaporated to small volume. The complex was precipitated with cold absolute ethanol, washed with 9∶1 acetone–water solution until free from chloride and dried under vacuum. Calc. for C14H21ClKN2O9Ru: C, 31.32; H, 4.19; N, 5.14. Found: C, 31.20; H, 4.30; N, 5.20%. IR (KBr): 3450 (OH); 1750 (COOH); 1655 (COO− asym); 1375 (COO− sym); 1220 (CO); 885, 685 (OCO def); 520 cm−1 (M–N). UV-vis, λ/nm (ε/M−1 cm−1): 240 (2430); 285 (1640) and 370 (700).
K[Ru(H2dtpa)Cl]
8..
This complex was isolated from K2[RuCl5(OH2)] and H5dtpa (0.403 g, 1.02 mmol) in 20 mL of HClO4 by the procedure described in the case of 7. Calc. for C16H20ClKN3O12Ru: C, 29.82; H, 3.49; N, 7.34. Found: C, 29.7; H, 3.56; N, 7.43%. IR (KBr): 3560 (OH); 1750 (COOH); 1655 (COO− asym); 1375 (COO− sym); 1280 (CO); 885, 685 (OCO def); 525 cm−1 (M–N). UV-vis, λ/nm (ε/M−1 cm−1): 235 (2235); 280 (1560) and 370 (375).
Instrumentation
Perkin-Elmer Series II-2400, CHNS/O Analyzer for C, H, N-data; Bio-Rad FT-40 spectrometer coupled to a SP-3200 computer for IR; Shimadzu UV-vis NIR scanning spectrophotometer UV-3101 PC for absorption spectra. An Adair Dutt digital pH meter (±0.01 pH) was used to record the pH of all experimental solutions other than those in potentiometry.
Electrochemical measurements were performed on EG&G Princeton Applied Research (PARC) instruments. A model PAR 174A Polarographic Analyzer and PAR 175 Universal Programmer coupled to a high precision Houston X-Y recorder were used to record sampled dc polarograms and cyclic voltammograms. A three electrode assembly, PAR 303 SMDE/HMDE comprising a dropping (DME, 3.85 mg s−1)/hanging (HMDE, 0.021 cm2) mercury drop working, platinum wire auxiliary and SCE reference electrodes was employed.
The controlled potential coulometry was performed on a EG&G PAR model 173 Potentiostat/Galvanostat coupled to a model 179 digital coulometer provided with a three electrode cell assembly. The cell consisted of a mercury pool (4 cm convex diameter) working electrode in the main compartment along with a platinum mesh as counter electrode separated by a glass frit, SCE reference electrode and a glass disc agitator. An Orion 940 Ion Analyzer equipped with a model 9512 ammonia sensing membrane electrode (sensitive to 10−12 M) was used for the estimation of ammonia in the electrolysed solutions.
Analytical methods
All acid–base titrations were conducted in a jacketed glass-walled cell of about 150 mL capacity, connected to a Metrohm Swiss model 682 Auto Titroprocessor coupled with a 665 Dosimat and E-649 magnetic stirrer, through a combined glass-reference electrode. The cell also had inlet and outlet tubes for passing Ar gas through the solution. The electrode system was calibrated in terms of H+ concentration by means of a titration of HCl solution with NaOH29 in 0.1 M Na2SO4 at 25 ± 0.1 °C. The experimental data ranged over pH values between 1.6 and 11.2.
In a typical experiment, a weighed quantity of a complex 1–8, N2H5HSO4 or N2H4PhCl (1 mM) was dissolved in 50 mL of 0.1 M Na2SO4 and titrated independently against carbonate free (0.1 M) NaOH solution in the region 2 < pH < 10 under Ar at 25 °C. Similar titrations were conducted in the presence of 1 equivalent of N2H5HSO4/N2H4PhCl. The resulting data were used to calculate the acid dissociation (pKa) constants corresponding to N2H4X+, 1–8 and adducts of both, and the metal hydrolysis (pKOH) constants of the last two systems. The titration data at pH > 10 in the case of 1–8 and at pH > 9.0 in the case of N2H4X+ adducts were not considered in the present investigations. The acid dissociation constants of N2H4X+ and the unco-ordinated COOH/CH2CH2OH in complexes 1–8 and in their 1∶1 adducts of N2H4X+, where the dissociation of protons takes place in well defined buffer regions, were evaluated29 with the help of Kw (1.008 × 10−14 at 25 °C) and suitable mass and charge balance equations fitting to the generalized eqn. (1). Charges on the species are omitted for the sake of clarity. The stepwise first, second or third metal hydrolyses of 1–8 and their N2H4X+ adducts were considered according to reactions (2). The hydrolysis constants falling in the pH range 2 < pH < 10 were calculated with the help of suitable mass balance equations and Kw at 25 °C.
| |
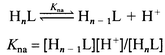
|
(1)
|
| |
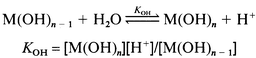
|
(2)
|
A graphical method30 was used to calculate the dissociation constants, in the case of simultaneous dissociation of two protons (either by the participation of COOH/CH2CH2OH and/or in the combination of dissociation of N2H4X+ proton and metal hydrolysis) described by the generalized eqn. (3) or combined eqns. (1) and (2), respectively.
| |
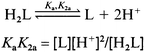
|
(3)
|
The absorption spectral studies on complexes 1–8 (1 mM) in the absence and in the presence of various concentrations of N2H5+ or N2H4Ph+ (1–100 mM) were performed at pH 2.8 in a 10 mm quartz cuvette and the spectra were recorded in the wavelength range between 200 and 700 nm at 25 ± 0.1 °C.
The voltammetric responses were recorded with a weighed quantity of complex (1 mM) in the absence or presence of a known concentration (0.1–100 mM) of substrate (N2H5+/N2H4Ph+) in 10 mL electrolyte solution at the desired pH under Ar at 25 °C. The ivs.E plots were recorded between +0.2 and −0.8 V (vs. SCE) at the sweep rates 10 mV s−1 in sampled dc and 20–500 mV s−1 in cyclic voltammetry (CV). The effect of pH on the diffusion current (id) and half-wave potentials (E1/2) was studied in the range 1–5. All the solutions were deaerated at least 15 min prior to measurements and the experiments were maintained at 25 ± 0.1 °C unless otherwise stated. The criteria for the electrochemical reversibility and diffusion current were established by following the reported techniques.31,32
Constant potential electrolysis was performed in 25 mL of deaerated buffer solution having the desired pH (1.9/2.8) containing 1 mmol of complex (1–8) and 100 mmol of substrate at predetermined potential for at least 10 h under Ar at 25 ± 0.1 °C. The ammonia produced during the reaction was estimated every hour21 while the aniline produced concomitantly with ammonia was tested qualitatively using vanadium(V) salt.33
Results and discussion
Interaction of N2H4X+ with the complexes 1–8
Potentiometry..
Complexes 1–8 were independently titrated against standard sodium hydroxide in the absence and in the presence of one equivalent of N2H4X+. The representative titration data thus obtained with 1 are presented in Fig. 1. Complex 1 (Fig. 1(a)) showed three titratable protons dissociating in three independent buffer regions, two in acidic and one in basic pH by the metal hydrolysis. Further hydrolysis of 1 occurring beyond a > 3 resulted in a dark brown solution which may be due to its decomposition and hence was not considered here. Similarly, complexes 2 and 3 showed three buffer regions caused by metal hydrolyses at 2 < pH < 10 with two ill defined inflections at a = 1 and 2. On the other hand, 4–7 showed the stepwise liberation of two protons in two well defined buffer regions 0 < a < 1 and 1 < a < 2. Of the two, the first buffer region is assigned to the dissociation of an unco-ordinated CO2H proton in the case of complexes 5–7 and may be the OH (CH2CH2OH) proton in the case of 4 while the second buffer region is considered for the metal hydrolysis.33,34 On the contrary, the complex 8 showed one buffer region 0 < a < 2 in acidic pH followed by another buffer region 2 < a < 3 in basic pH. The first buffer region is assigned to the simultaneous liberation of two protons on the dtpa ligand, the second to metal hydrolysis.
![Potentiometric titration curve of (a) K2[Ru(imda)Cl3]·2H2O, 1 mM; (b) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H5+, 1 mM; (c) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H4Ph+, 1 mM at 25 °C. I = 0.1 M (Na2SO4), a = mols of base added per mol of complex or N2H4X+.](/image/article/2000/DT/a908338c/a908338c-f1.gif) |
| | Fig. 1 Potentiometric titration curve of (a) K2[Ru(imda)Cl3]·2H2O, 1 mM; (b) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H5+, 1 mM; (c) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H4Ph+, 1 mM at 25 °C. I = 0.1 M (Na2SO4), a = mols of base added per mol of complex or N2H4X+.
| |
The metal hydrolysis steps, three in the case of complex 1, the first two in the case of 2 and 3, the single one in the case of 4–6 and 8, and the first one in the case of 7, are accounted for by the rapid substitution of chloride ions by H2O as verified kinetically in the case of 4–6, and the liberation of titratable protons following its dissociation.26,35 The third metal hydrolysis step in the case of 2 and 3 could be explained if one of the loosely bound carboxylate groups of the tetradentate himda and nta is substituted by H2O or the liberation of OH (CH2CH2OH) proton on tridentate himda in 2. The acid (pKa) and metal hydrolysis (pKOH) constants were calculated with the help of eqns. (1)–(3) and are given in Table 1. These showed that the complexes 1–3 are more hydrolysable because the chelation capability of imda (tri), himda (tri or tetra) and nta (tetra) ligands is low. All other complexes, 4–8, showed a single hydrolysis step which occurs almost at neutral pH. This implies that all these complexes have a potential site available to another ligand for co-ordination/substitution, though the number of co-ordinating sites in the aminopolycarboxylic acid ligand varies from 5 to 8. The present data also revealed that only five co-ordination sites on the hexacovalent ruthenium are used by all penta- or higher poly-dentate aminopolycarboxylic acids in the process of bond formation. The hydrolytic tendency of the metal increased in the order 8 < 4 < 5 < 6 < 7 < 2 < 3 < 1.
Table 1 Acid dissociation (pKa) and metal hydrolysis (pKOH) constants of complexes 1–8 at 25 °C, I = 0.1 M (Na2SO4)
| |
pKa |
pKOH |
| Complex |
CO2H |
CH2CH2OH |
First |
Second |
Third |
| Values given in parentheses were evaluated by a graphical method. |
|
1
|
— |
— |
2.42 ± 0.02 |
5.78 ± 0.03 |
8.86 ± 0.03 |
|
2
|
— |
— |
2.86 ± 0.02 |
5.72 ± 0.02 |
8.53 ± 0.03 |
|
3
|
— |
— |
2.47 ± 0.01 |
4.80 ± 0.03 |
9.62 ± 0.03 |
|
4
|
— |
4.82 ± 0.03 |
7.96 ± 0.03 |
— |
— |
|
5
|
2.34 ± 0.02 |
— |
7.64 ± 0.02 |
— |
— |
|
6
|
2.38 ± 0.02 |
— |
7.60 ± 0.02 |
— |
— |
|
7
|
2.55 ± 0.03 |
— |
6.20 ± 0.03 |
— |
— |
|
8
|
(2.58, 3.40) |
— |
8.28 ± 0.02 |
— |
— |
The titration data of N2H5HSO4 showed two buffer regions separated by an inflection at a = 1. The buffer region 0 < a < 1 is considered for the neutralization of the strong acidic proton due to the HSO4− group, since the N2H6+ (Ka 11)11 does not form inder the present experimental conditions. The buffer region corresponding to this proton was ignored in all other studies presented below and hence not included in Fig. 1(b), but considered in the evaluation of relevant constants. The second buffer region 1 < a < 2 is assigned to the neutralization of proton in the equilibrium N2H5+ ⇌ N2H4 + H+. Similarly, the titration data of N2H4PhCl contained a single buffer region 0 < a < 1, representing the equilibrium N2H4Ph+ ⇌ N2H3Ph + H+.
The titration curves of complexes 1–8 in the presence of N2H5+ possessed buffer regions, four in the case of 1 (Fig. 1(b)) and three in the case of 2–8. The last buffer region (fourth in the case of 1, and third in the case of other complexes), where RuIII–N2H5+ species were decomposed to give ruthenium(II) complexes, as found by sampled dc at pH > 9, was not considered in the present study. The third buffer region in the case of 1 and the second in the case of 2–8, which were assigned earlier to metal hydrolysis in the absence of N2H5+, are assigned to the equilibrium N2H5+ ⇌ N2H4 + H+ in the vicinity of the metal co-ordination sphere. The first and second buffer regions in the case of 1 and the first in the case of 2 and 3 are assigned to the metal hydrolysis, while the first in the case of 4–7 and the first and second buffer regions in the case of 8 are assumed for the deprotonation of the CO2H/CH2CH2OH group on the aminopolycarboxylic acid as found above in the absence of N2H5+.
The potentiometric data obtained in the presence of N2H4PhCl showed two buffer regions: a = 0–1, 1–3 in the case of complex 1 (Fig. 1(c)), a = 0–1, 1–2 in the case of 2, 3 and 5–7 and a = 0–2, 2–3 in the case of 8, while one buffer region a = 0–2 in the case of 4. It was noticed that the hydrolysis steps, third in the case of 1, second in the case of 2 and 3, and first in the case of 5–8 which were observed in the absence of N2H4Ph+, were not seen. Thus, the deprotonation of N2H4Ph+ was assumed in the buffer region a = 1–3 in the presence of 1 along with the second metal hydrolysis, 1–2 in the presence of 2, 3, 5–7, 0–2 in the case of 4 together with the deprotonation of the OH (CH2CH2OH) proton and 2–3 in the case of 8.
The acid dissociation constants and metal hydrolysis constants corresponding to free N2H4X+ and its adducts with complexes 1–8 were calculated with the help of eqns. (1)–(3) and presented in Table 2. The values of Ka (CO2H/CH2CH2OH) and KOH are closely comparable to the corresponding values seen in Table 1. Moreover, the values obtained for N2H4X+ ⇌ N2H3X + H+ in the presence of 1–8 are in close agreement with those obtained in their absence. However, the Ka value is reduced to only 0.38–1.0 logarithmic units in the case of N2H5+ and negligibly affected in the case of N2H4Ph+ after their interaction with the ruthenium complex. On the basis of these observations together with the disappearance of metal hydrolysis steps (third in the case of 1, second in the of 2 and 3, and first in the other complexes as seen in Table 1) in the presence of N2H4X+, the co-ordination of N2H4X+ to ruthenium occurs at the vacant site where the poorly dissociable water was attached. Further, the small changes in pKa values of the N2H4X+ ligand after its interaction with ruthenium suggests that its co-ordination takes place through the weakly basic nitrogen (NH2 of N2H5+ or NHPh of N2H4Ph+) leaving the other nitrogen in the protonated form, which is further confirmed by the voltammetric data presented at pH < 3.0 in the latter part of this paper.
Table 2 Acid dissociation (pKa) and metal hydrolysis (pKOH) constants of N2H4X+ and their adducts with complexes 1–8 at 25 °C, I = 0.1 M (Na2SO4)
| Adduct |
pKa |
pKOH |
| N2H4X+ |
Complex |
N2H4X+ |
CO2H/CH2CH2OH |
First |
Second |
| Values given in parentheses were evaluated by a graphical method. |
| N2H5+ |
— |
8.05 ± 0.01 |
— |
— |
— |
| |
1
|
7.67 ± 0.02 |
— |
2.43 ± 0.02 |
5.82 ± 0.03 |
| |
2
|
7.42 ± 0.03 |
— |
2.82 ± 0.02 |
|
| |
3
|
7.36 ± 0.02 |
|
2.45 ± 0.02 |
— |
| |
4
|
7.65 ± 0.02 |
4.83 ± 0.02 |
— |
— |
| |
5
|
7.52 ± 0.02 |
2.40 ± 0.02 |
— |
— |
| |
6
|
7.28 ± 0.03 |
2.42 ± 0.02 |
— |
— |
| |
7
|
7.15 ± 0.02 |
2.58 ± 0.02 |
— |
— |
| |
8
|
7.30 ± 0.03 |
(2.60, 3.43) |
— |
— |
| N2H4Ph+ |
— |
5.27 ± 0.02 |
— |
— |
— |
| |
1
|
(5.24) |
— |
2.45 ± 0.02 |
(5.84) |
| |
2
|
5.25 ± 0.02 |
— |
2.89 ± 0.03 |
— |
| |
3
|
5.34 ± 0.02 |
— |
2.50 ± 0.03 |
— |
| |
4
|
(5.32) |
(4.62) |
— |
— |
| |
5
|
5.20 ± 0.02 |
2.35 ± 0.02 |
— |
— |
| |
6
|
5.22 ± 0.02 |
2.41 ± 0.02 |
— |
— |
| |
7
|
5.31 ± 0.02 |
2.60 ± 0.02 |
— |
— |
| |
8
|
5.30 ± 0.02 |
(2.64, 3.49) |
— |
— |
Absorption spectra..
The electronic absorption spectra of complexes 1–8 were recorded under identical conditions in the absence and presence of N2H4X+ salts at different compositions at pH 2.8 to verify the interaction of the latter (N2H4X+) through the weakly basic nitrogen with ruthenium. In the absence of N2H4X+ the ligand charge transfer between 240 and 250 nm and the ligand to metal charge transfer bands in the regions 280–290 and 360–370 nm were observed for all complexes. The absorption band at 360 nm, characteristic of a Ru–OH2 bond,36 was reduced in intensity on introduction of N2H5+ due to the replacement of bound water molecule by the latter. In the case of addition of N2H4Ph+ two new absorption bands at 400–450 and 540 nm appeared rapidly. The band at 540 nm did not occur in the case of complexes 7 and 8. Concomitantly, the yellow colour of these complexes instantly changed to reddish brown accounting for the formation of N2H4Ph+ adducts in solution. Further, plots of change in absorbance (ΔA) vs. the ratio [N2H4X+]∶[complex] confirmed the formation of 1∶1 N2H4X+ adducts as suggested above with 1–8. Fig. 2 shows the typical spectral changes observed with 4 in the absence and in the presence of N2H4X+ (1 mM) at pH 2.8. The spectral features observed with all complexes 1–8 are summarized in Table 3.
Table 3 Absorption spectral data of complexes 1–8 in the absence and in the presence of N2H4X+ at pH 2.8
| |
λ
max/nm (εmax/M−1 cm−1) |
| Complex |
Absence |
Presence of N2H5+ |
Presence of N2H4Ph+ |
|
1
|
240(2270); 285(1400); 365(445) |
240(2240); 285(1340) |
420(1070); 540(350) |
|
2
|
245(2595); 285(2360); 360(640) |
245(2370); 285(2320) |
425(1450); 540(350) |
|
3
|
245(2330); 285(2200); 360(735) |
245(2290); 285(2120) |
420(1360); 540(350) |
|
4
|
240(2270); 285(1400); 360(445) |
240(2240); 285(1375) |
400(1400); 540(260) |
|
5
|
245(2600); 285(2370); 360(640) |
245(2570); 285(2360) |
420(2250); 540(480) |
|
6
|
245(2565); 285(2420); 360(630) |
245(2560); 285(2380) |
400(1350); 540(130) |
|
7
|
240(2430); 285(1630); 360(700) |
240(2390); 285(1580) |
430(1230) |
|
8
|
235(2240); 280(1560); 360(375) |
235(2230); 280(1540) |
450(1270) |
![Absorption spectra of (a) K[Ru(hedta)Cl], 1 mM; (b) K[Ru(hedta)Cl], 1 mM and N2H5+, 1 mM; (c) K[Ru(hedta)Cl], 1 mM and N2H4Ph+, 1 mM. In all cases the pH was 2.8.](/image/article/2000/DT/a908338c/a908338c-f2.gif) |
| | Fig. 2 Absorption spectra of (a) K[Ru(hedta)Cl], 1 mM; (b) K[Ru(hedta)Cl], 1 mM and N2H5+, 1 mM; (c) K[Ru(hedta)Cl], 1 mM and N2H4Ph+, 1 mM. In all cases the pH was 2.8.
| |
Voltammetry..
Sampled dc polarograms of complexes 1–8 were recorded in the absence and in the presence of various equivalents of N2H4X+. In the absence, each complex exhibited a well defined one-electron cathodic wave in the potential range between −0.150 and −0.400 V (E1/4 − E3/4 55–65 mV; I (diffusion current constant) 1.2–1.5) vs. SCE assignable to the RuIII → RuII process. The height of this wave proportionately enhanced with increase in complex concentration, while the plots Ede (potential at the electrode) vs. log [i/(id − i)] were linear with a slope of about 70 mV, indicating the electrode reactions are one-electron, diffusion controlled and quasi-reversible processes. The wave shifted to a measurable extent (60 mV per pH unit) in the case of 1–3, less in the case of other complexes with the change in pH between 1.0 and 5.0. The polarogram of 1 representing this one-electron wave is shown in Fig. 3(a) while the half-wave potential (E1/2) data of all the complexes 1–8 are listed in Table 4. It decreased in the order 8 > 5 > 4 > 6 > 7 > 3 > 2 > 1 which is in good agreement with the order of sigma basicity of the co-ordinated aminopolycarboxylic acid.37 However, it is noted that the E1/2 values for 1–3 are relatively more negative than those of other complexes. This may be attributed to the strong hydrolytic tendency of these complexes when dissolved in aqueous solution. Accordingly, the hydrolytic tendency of these complexes may be given as 3 < 2 < 1. On the other hand, the lower E1/2 value for complex 8 than those of 4–7 may be accounted for by its destabilization due to steric repulsion because of the large size of dtpa, despite its large sigma basicity.
Table 4 Half wave potentials of the RuIII → RuII wave and those of complexes 1–8 observed in the presence of 100 equivalents of N2H4X+ at pH 2.8
| |
−E1/2/V vs. SCE |
| Complex |
Absence |
Presence of N2H5+ |
Presence of N2H4Ph+ |
|
1
|
0.372 |
0.346, 0.475 |
0.360, 0.482 |
|
2
|
0.364 |
0.328, 0.450 |
0.340, 0.462 |
|
3
|
0.354 |
0.300, 0.440 |
0.322, 0.456 |
|
4
|
0.248 |
0.030, 0.256 |
0.165, 0.248, 0.335 |
|
5
|
0.238 |
0.025, 0.230 |
0.150, 0.225, 0.346 |
|
6
|
0.251 |
0.034, 0.250 |
0.156, 0.235, 0.340 |
|
7
|
0.286 |
0.045, 0.230 |
0.168, 0.260, 0.386 |
|
8
|
0.165 |
0.028, 0.250 |
0.154, 0.250, 0.395 |
![Sampled dc polarogram of (a) K2[Ru(imda)Cl3]·2H2O, 1 mM; (b) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H5+, 100 mM; (c) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H4Ph+, 100 mM. The pH in each case was maintained at 2.8.](/image/article/2000/DT/a908338c/a908338c-f3.gif) |
| | Fig. 3 Sampled dc polarogram of (a) K2[Ru(imda)Cl3]·2H2O, 1 mM; (b) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H5+, 100 mM; (c) K2[Ru(imda)Cl3]·2H2O, 1 mM and N2H4Ph+, 100 mM. The pH in each case was maintained at 2.8.
| |
When 0.1–5.0 equivalents of N2H5+ were introduced into the complex solution the RuIII → RuII wave was enhanced while its plateau extended to anodic potentials in cases of complexes 1–8. As a result, the E1/2 value apparently shifted to anodic potentials. A plot of the enhanced current increased linearly as the number of equivalents of added N2H5+ approached unity and remained constant thereafter indicating the instant formation of monomeric N2H5+ complexes in acidic solutions, as suggested above. The plateau of the enhanced wave gradually split into two as [N2H5+] reached twenty times more than that of the complex and the wave finally appeared as two distinguishable waves (w1 and w2) at all other compositions of N2H5+. The polarographic responses of 1 in the presence of 100 equivalents of N2H5+ are shown in Fig. 3(b). In all cases, the wave w1 (E1/4 − E3/4 32 mV; I 1.9) shifted anodically with the increase in [N2H5+] while the wave w2 (E1/4 − E3/4 65 mV; I 1.0) remained steady. On the other hand, the diffusion currents of w1 and w2 were directly proportional to [complex]. The plots Edevs. log [i/(id − i)] for w1 and w2 were linear with slopes of 33 and 68 mV, respectively. The diffusion current (id) of the composite wave or of the wave w1 or w2 was maximum in the pH range 2.5–3.5 and somewhat reduced when the ionic strength was raised beyond 0.5 M. Besides, the wave w1 shifted negligibly with increase in pH between 1 and 4. The E1/2 data (Table 4) revealed that the wave w1 is 0.02–0.05 V less negative in the case of 1–3, while it is 0.13–0.22 V less negative in the case of 4–8 compared to that of the corresponding one-electron RuIII → RuII wave observed before the addition of N2H5+. Similarly, the wave w2 is about 0.09–0.10 V more negative in the case of 1–3 and 8 and similar in the case of 4–7 to that of the initial one-electron RuIII → RuII wave. The wave w1 in the case of 1–3 is more negative than that of the complexes 4–7. The E1/2 of w1 is correlated with ΣpKa of the primary ligand,37 aminopolycarboxylic acid. The large negative E1/2 for complexes 1–3 is responsible for their hydrolytic tendency, while the negligible dependence of E1/2 in complexes 4–8 shows that there is no considerable change in the redox properties of the metal with the increase in sigma basicity of the aminopolycarboxylic acid. Probably, this could be due to identical binding of penta-, hexa- or hepta-dentate aminopolycarboxylic acids to ruthenium through the same number of co-ordinating groups.
The polarographic responses of complex 1 in the presence of 0.1–100 equivalents of N2H4Ph+ were probed. The response in 100 equivalents of N2H4Ph+ is depicted in Fig. 3(c). It displayed two closely separated waves (w1 and w2) at potentials close to that of the RuIII → RuII wave with a doubled diffusion current. The responses in the presence of other concentrations (0.1–100 equivalent of N2H4Ph+) overlapped those in Fig. 3(c) except for a negligible negative shift in E1/2 and a minor change in the overall diffusion current. Complexes 2 and 3 also exhibited two cathodic waves replacing the RuIII → RuII wave while the other complexes (4–8) showed an additional low intensity wave poorly resolved from w1 and w2. The E1/2 data pertaining to these waves are summarized in Table 4. The measured data (E1/4 − E3/4 34 mV and I 2.0 for w1; 65 mV and 0.9 for w2) indicated w1 to be a two-electron process while w2 and the additional wave are one-electron processes. This was further substantiated by plots of Edevs. log [i/(id − i)] which were linear having slopes of 32–35 mV for w1 and 67–70 mV for both w2 and the additional wave. The overall diffusion current at −0.5 V was proportional to [1] while the enhanced current (Δid) was a maximum in the pH range 2.0 to 3.5. The data in Table 4 revealed that, for a given complex, the E1/2 of both w1 and w2 are more negative with N2H4Ph+ than with N2H5+, indicating that the former hydrazine binds to ruthenium more strongly than the latter can through a sigma bond. This may due to the effect of phenyl ring substitution on the co-ordinating nitrogen in the former hydrazine. The E1/2 values of w1 obtained with complexes 1–8 in the presence of N2H4Ph+ are correlated with ΣpKa of the aminopolycarboxylic acid37 attached to ruthenium. Again, the large negative values in the case of 1–3 and negligible dependence of E1/2 in the case of 4–8 are accounted for by the reasons given before.
The CV responses of complexes 1–8 in the absence and in the presence of N2H4X+ were run subsequently for the above solutions. The results in the absence and in the presence of 100 equivalents of N2H4X+ are discussed below. All the complexes, in the absence of N2H4X+, yielded a pair of cathodic and anodic peaks corresponding to the quasi-reversible RuIII–RuII couple. Fig. 4(a) shows a typical cyclic voltammogram of 4, which peaked at −0.320 V in the forward scan and at −0.252 V in the reverse scan. The peak separation which varied between 70 and 65 mV at 0.1 V s−1 increased with increase in scan speed, while the peak width Ep/2 − Ep tended to 62–65 mV which is close to the theoretical value for a one-electron process. On the other hand, the peak currents increased linearly with increase in the square root of the scan speed, while the peak to peak ratio (ipa∶ipc) was less than unity.
![Cyclic voltammograms of (a) K[Ru(Hcdta)Cl]·H2O, 1 mM; (b) K[Ru(Hcdta)Cl]·H2O, 1 mM and N2H5+, 100 mM; (c) K[Ru(Hcdta)Cl]·H2O, 1 mM and N2H4Ph+, 100 mM. The pH in each case was maintained at 2.8.](/image/article/2000/DT/a908338c/a908338c-f4.gif) |
| | Fig. 4 Cyclic voltammograms of (a) K[Ru(Hcdta)Cl]·H2O, 1 mM; (b) K[Ru(Hcdta)Cl]·H2O, 1 mM and N2H5+, 100 mM; (c) K[Ru(Hcdta)Cl]·H2O, 1 mM and N2H4Ph+, 100 mM. The pH in each case was maintained at 2.8.
| |
All the complexes, in the presence of 100 equivalents of N2H5+, exhibited two well resolved cathodic peaks (p1 and p2 corresponding to the polarographic waves w1 and w2, respectively) and a single anodic peak (p3, counterpart of p2). The anodic counterpart of p1 did not appear under the present experimental conditions. Representative CV responses of complex 4 are shown in Fig. 4(b). The complexes 1–3 exhibited two well resolved cathodic peaks (p1 and p2) in the presence of 100 equivalents of N2H4Ph+. Contrarily, they showed a single anodic peak (p4) as counterpart of p1 and no peak for p2. Complexes 4–8 behaved similarly in the presence of 100 equivalents of N2H4Ph+. Additionally, they showed a well defined cathodic peak and its counterpart (Fig. 4(c)) corresponding to the additional wave observed in sampled dc between w1 and w2. The peak analyses data for p1 and p2, confirmed them to be a two- and one-electron reduction process, respectively. Peak p1 shifted anodically with increase in [N2H4X+] but cathodically with increase in scan speed, but plots of ipcvs.ν1/2 were linear for both p1 and p2.
Mechanism of electrode reactions
The complexes 1–8 were reduced in solution in the absence and in the presence of one equivalent of N2H4X+ at pH 1.9 and 2.8 by holding the potential at a mercury pool cathode at 0.1 V away from the E1/2 of RuIII → RuII wave as given in Table 4. In the absence, all the complexes consumed one faraday of charge and produced the corresponding ruthenium(II) complexes. In the presence of N2H4X+ all the complexes consumed three faradays. The electrolysed solutions contained an ammonia concentration two times that of [N2H5+] but equal to that of [N2H4Ph+] taken initially. On the basis of these data and the data in Figs. 3 and 4, it is proposed that the RuIII–N2H4X+ complexes take two electrons from the electrode at w1 and produce RuI–N2H4X+ species (reaction (4), charges on the species are omitted for clarity). These ruthenium(I) species are less stable and hence rapidly decompose as shown in reaction (5) to RuIII–NH2X and one mole of NH3 by abstracting one equivalent of H+ from the medium. The species RuIII–NH2X are reduced by one electron in a subsequent step at w2 (reactions (6) and (10)) to give RuII–NH2X which on aquation produces the corresponding RuII–H2O species and NH2X (reactions (8) and (12)), or hydrolyse to RuIII–OH2 producing an equivalent amount of NH2X (reactions (7) and (11)). The RuIII–OH2 species thus produced may be reduced along with RuIII–NH2X at w2 in the case of N2H5+ (reaction (9)) or at potentials where a new reduction step appeared between w1 and w2 in the case of N2H4Ph+ (reaction (13)). The RuII–NH2Ph complexes produced at w1 are not as stable as the corresponding RuIII–NH3 due to the presence of the Ph group and hence these species and their reduced forms RuII–NH2Ph quickly hydrolyse to the corresponding aqua species and NH2Ph (reactions (11) and (12)). This may be the reason for the irreversible nature of wave w2 and reversible nature of the poorly resolved new wave between w1 and w2.
| | |
RuIII–N2H4X+ + 2e− ⇌ RuI–N2H4X+
|
(4)
|
| | |
RuI–N2H4X+ + H+ → RuIIINH2X + NH3
|
(5)
|
When X = H
| | |
RuIII–NH3 + e− ⇌ RuII–NH3
|
(6)
|
| | |
RuIII–NH3 + H2O → RuIII–H2O + NH3
|
(7)
|
| | |
RuII–NH3 + H2O → RuII–H2O + NH3
|
(8)
|
| | |
RuIII–H2O + e− ⇌ RuII–H2O
|
(9)
|
When X = Ph
| | |
RuIII–NH2Ph + e− ⇌ RuII–NH2Ph
|
(10)
|
| | |
RuIII–NH2Ph + H2O → RuIII–H2O + NH2Ph
|
(11)
|
| | |
RuII–NH2Ph + H2O → RuII–H2O + NH2Ph
|
(12)
|
| | |
RuIII–H2O + e− ⇌ RuII–H2O
|
(13)
|
Electrolytic reduction of N2H4X+ in the presence of complexes 1–8
The hydrazines N2H4X+ were electrolytically reduced at pH 1.9 and 2.8 under Ar by constant potential coulometry by holding the potential at 50 mV away from the corresponding E1/2 of w1 given in Table 5 in order to measure the catalytic ability of the ruthenium as a function of the sigma basicity of the aminopolycarboxylic acid on the reduction of the N–N bond in N2H4X+. In all cases the constant reduction of N2H4X+ to NH3 and NH2X occurred, eqn. (14), justifying the wave w1 as a multi-electron process.
Table 5 Turnover number, moles of NH3 produced per mol of complex per hour, during the reduction of N2H4X+
| |
Turnover number |
| |
N2H5+ |
N2H4Ph+ |
| Complex |
Potential/V |
pH 1.9 |
pH 2.8 |
Potential/V |
pH 1.9 |
pH 2.8 |
|
1
|
−0.350 |
1.44 |
2.54 |
−0.400 |
0.94 |
1.10 |
|
2
|
−0.200 |
1.76 |
4.12 |
−0.300 |
1.02 |
1.76 |
|
3
|
−0.350 |
2.98 |
6.06 |
−0.400 |
1.78 |
2.98 |
|
4
|
−0.200 |
3.75 |
7.18 |
−0.250 |
1.62 |
2.95 |
|
5
|
−0.050 |
9.50 |
18.40 |
−0.200 |
2.85 |
5.98 |
|
6
|
−0.100 |
7.96 |
14.84 |
−0.220 |
2.68 |
5.32 |
|
7
|
−0.175 |
4.46 |
8.98 |
−0.300 |
2.17 |
3.84 |
|
8
|
−0.100 |
1.34 |
2.95 |
−0.230 |
0.92 |
1.27 |
| |
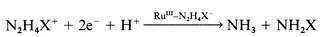
|
(14)
|
At both pH studied, the number of mols of NH3 produced per mol of complex was linear, in all cases, as seen in Fig. 5 for N2H5+ reduction. The turnover number and mols of NH3 formed per mol of complex per hour are given in Table 5. For a given substrate, the turnover number at pH 2.8 is nearly double that at pH 1.9. The observed turnover number of NH3 obtained with N2H4Ph+ is less than half of that achieved with N2H5+. However, the coulombic efficiency in all these cases was almost 100%. The solution at the end of electrolysis showed the same voltammetric features as described above and produced NH3 at the same turnover rate when an additional amount of N2H4X+ was added to the electrolysed solution and the electrolysis continued after adjusting the pH to the initial value. Thus, the catalytic efficiency of the metal was shown to be intact during the reduction cycle.
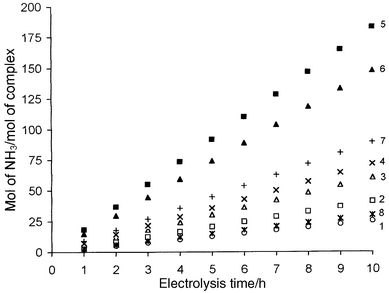 |
| | Fig. 5 Plots of mols of NH3 produced per mol of complex (1–8) during the reduction of N2H5+ at pH 2.8 vs. electrolysis time.
| |
The turnover rate is correlated in Fig. 6 with the sigma basicity of the polyaminopolycarboxylic acid. The data revealed that the turnover rate increases with the increase in sigma basicity of the aminopolycarboxylic acid to nearly ΣpKa = 22 and then decreases thereafter. With the given hydrazine N2H5+/N2H4Ph+, it increases in the order 1 < 2 < 3 < 4 < 5 ≈ 6 as the sigma basicity of the aminopolycarboxylic acid increases in the order 2 < 1 < 3 < 4 < 5 < 6. The lower turnover rate for complex 1 than that of 2 is explained by its high hydrolytic tendency. The decrease in the turnover rate with 5–8 with the increase in the sigma basicity may be related to the structural constraints developed within the aminopolycarboxylic acid due to increase in size, affecting the overall catalytic ability of the ruthenium.
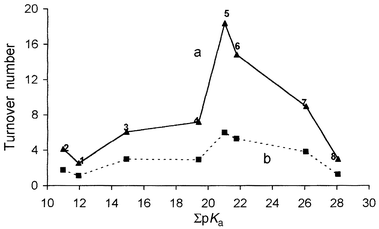 |
| | Fig. 6 Plot of turnover number of NH3 produced by the reduction of N2H4X+vs.
ΣpKa of the aminopolycarboxylic acid: (a) N2H5+, (b) N2H4Ph+.
| |
Conclusion
Ruthenium(III)–aminopolycarboxylic acid complexes of tri- (imda, himda), tetra- (himda, nta), penta- (hedtra), hexa- (edta, pdta and cdta) and octa- (dtpa) dentate ligands were prepared and characterized. Interaction of these complexes with hydrazines N2H4X+ (X = H or Ph) was investigated by pH-metry, voltammetry and spectrophotometry. The results confirmed that these complexes rapidly form monomeric adducts with N2H4X+ by its substitution at the less hydrolysable metal co-ordination site in acidic media. These adducts readily accept two electrons to form the corresponding RuI–N2H4X+ which on decomposition give one mol of ammonia and an electroactive species which in a subsequent chemical step gives one mol of NH2X. Employing these hydrazinium adducts, N2H4X+ were reduced electrolytically. Ammonia and/or amine was obtained in catalytic amounts with nearly 100% coulombic efficiency. The turnover rate of ammonia produced per mol of complex per hour was evaluated and correlated with the sigma basicity (ΣpKa) of the aminopolycarboxylic acid. The data revealed that the turnover rate increased with increase in sigma basicity in the order imda < himda < nta < hedtra < edta ≈ pdta. Further increase in the sigma basicity of the aminopolycarboxylic acid reduced the catalytic ability of the ruthenium which is attributed to the development of complexity caused by the concomitant increase in size of the aminopolycarboxylic acid. The most probable chemical and charge transfer steps were suggested for the reactions at the electrode.
Acknowledgements
The authors are grateful to the Department of Science and Technology (No. SP/S1/F-12/93) New Delhi for supporting this work. R. P. is grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi for awarding a Senior Research Fellowship (31/28(22)/97 EMR-I and 31/28(29)/99 EMR-I).
References
- H. Dalton and L. E. Mortenson, Bacteriol. Rev., 1972, 21, 29 Search PubMed.
- J. Chatt, J. R. Dilworth and R. L. Richards, Chem. Rev., 1978, 78, 589 CrossRef CAS; P. Pelikan and R. Boca, Coord. Chem. Rev., 1984, 55, 55 CrossRef CAS.
- R. A. Handerson, G. J. Leigh and C. J. Pickett, Adv. Inorg. Chem. Radiochem., 1983, 27, 198 Search PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983 CrossRef CAS; J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965 CrossRef CAS.
- D. Sellmann and J. Sutter, Acc. Chem. Res., 1997, 30, 460 CrossRef CAS.
- B. E. Smith, M. C. Durrant, S. A. Fairhurst, C. A. Gormal, K. L. C. Gornburg, R. A. Henderson, S. K. Ibrahim, T. Le. Gall and C. J. Pickett, Coord. Chem. Rev., 1999, 185–186, 669 CrossRef CAS.
- R. N. F. Thorneley, R. R. Eady and D. J. Lowe, Nature (London), 1978, 272, 557 CAS.
- B. T. Heaton, C. Jacob and P. Page, Coord. Chem. Rev., 1996, 154, 193 CrossRef CAS and references therein..
- P. B. Hitchock, D. L. Hughes, M. J. Maguire, K. Marjani and R. L. Richards, J. Chem. Soc., Dalton Trans., 1997, 4747 RSC.
- J. V. Barkely, B. T. Heaton, C. Jacob, R. Mageswaran and J. T. Sampanthar, J. Chem. Soc., Dalton Trans., 1998, 697 RSC.
- T. Furuhashi, M. Kawano, Y. Koide, R. Somazawa and K. Matsumoto, Inorg. Chem., 1999, 38, 109 CrossRef CAS.
- R. R. Schrock, T. E. Glassman, M. G. Vale and M. Kol, J. Am. Chem. Soc., 1993, 115, 1760 CrossRef CAS.
- S. M. Malinak, K. D. Demadis and D. Coucouvanis, J. Am. Chem. Soc., 1995, 117, 3126 CrossRef CAS.
- T. A. George, D. N. Kurk and J. Redepenning, Polyhedron, 1996, 15, 2377 CrossRef CAS.
- G. N. Schrauzer, F. R. Robinson, E. L. Moorehead and J. M. Vickery, J. Am. Chem. Soc., 1976, 98, 2815 CrossRef CAS.
- Y. Hozumi, Y. Imasaka and T. Tanaka, Chem. Lett., 1983, 897 CAS.
- S. Cai and R. R. Schrock, Inorg. Chem., 1991, 30, 4106 CrossRef.
- S. Kuwata, Y. Mizobe and M. Hidai, Inorg. Chem., 1994, 33, 3619 CrossRef CAS.
- K. D. Demadis and D. Coucouvanis, Inorg. Chem., 1995, 34, 3658 CrossRef CAS; K. D. Demadis, S. M. Malinak and D. Coucouvanis, Inorg. Chem., 1996, 35, 4038 CrossRef CAS; S. M. Malinak, A. M. Simeonov, P. E. Mosier, C. E. Mckenna and D. Coucouvanis, J. Am. Chem. Soc., 1997, 119, 1662 CrossRef CAS.
- R. E. Palermo, R. E. Singh, J. K. Bashkin and R. H. Holm, J. Am. Chem. Soc., 1984, 106, 2600 CrossRef CAS.
- G. Ramachandraiah, J. Am. Chem. Soc., 1994, 116, 6733 CrossRef CAS.
- R. Prakash, B. Tyagi, D. Chatterjee and G. Ramachandraiah, Polyhedron, 1997, 12, 1235 CrossRef CAS.
- R. Prakash and G. Ramachandraiah, Stud. Surf. Sci. Catal., 1998, 113, 519 Search PubMed; D. Chatterjee, Coord. Chem. Rev., 1998, 168, 273 CrossRef CAS.
-
R. Prakash
and G. Ramachandraiah
, J. Mol. Catal. A, Chem.,
in the press..
- E. R. Mercer and R. R. Buckley, Inorg. Chem., 1965, 4, 1962.
- T. Matsubara and C. Creutz, Inorg. Chem., 1979, 18, 1956 CrossRef CAS;
J. Am. Chem. Soc., 1978, 100, 6255.
Search PubMed.
- A. A. Diamantis and J. V. Dubrawski, Inorg. Chem., 1981, 20, 1142 CrossRef CAS; M. M. Taqui Khan, A. Kumar and Z. Shirin, J. Chem. Res. (S), 1986, 5, 1003 Search PubMed.
- M. M. Taqui Khan, M. A. Moiz and A. Hussain, J. Coord. Chem., 1991, 23, 245 Search PubMed; M. M. Taqui Khan and R. M. Naik, J. Mol. Catal., 1989, 54, 139 CrossRef.
- M. M. Taqui Khan, A. Hussain, G. Ramachandraiah and M. A. Moiz, Inorg. Chem., 1986, 25, 3023 CrossRef; M. M. Taqui Khan and A. Hussain, Indian J. Chem., Sect. A, 1979, 19, 50 Search PubMed;
CRC Handbook of Chemistry and Physics, eds. R. C. Weast and M. J. Astle, CRC Press, Boca Raton, FL, 1979, p. D-168.
Search PubMed.
-
A. E. Martell
and M. Calvin
, in Chemistry of Metal Chelate Compounds, Prentice-Hall, New York, 1952.
Search PubMed.
-
L. Meites
, in Polarographic Techniques, Interscience, New York, 2nd edn., 1957, pp. 95–301.
Search PubMed.
-
A. J. Bard
and L. R. Faulkner
, in Electrochemical Methods Fundamentals and Applications, Wiley, New York, 1980.
Search PubMed.
-
V. N. Alexeyev
, in Qualitative Chemical Semimicro Analysis, ed. P. K. Agasyan, Mir Publishers, Moscow, 1975, p. 475.
Search PubMed.
- J. F. Endicott and H. Taube, Inorg. Chem., 1965, 4, 437 CrossRef CAS; K. Shimzu, T. Matsubara and G. P. Sato, Bull. Chem. Soc. Jpn., 1974, 47, 1651.
- P. D. Maria Mattioli and A. A. Luiz Oliveria, Polyhedron, 1987, 6, 603 CrossRef.
- H. Ogino, T. Watanabe and N. Tanaka, Inorg. Chem., 1975, 14, 2093 CrossRef CAS; H. Ogino, M. Shimura and N. Tanaka, Inorg. Chem., 1979, 18, 2497 CrossRef CAS.
-
A. E. Martell
and R. M. Smith
, in Critical Stability Constants, Plenum, New York, 1982, vols. 1–5.
Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.