Formation and gas phase fragmentation reactions of ligand substitution products of platinum(II) complexes via electrospray ionization tandem mass spectrometry†
Received
(in Cambridge, UK)
18th October 1999
, Accepted 4th November 1999
First published on 24th December 1999
Abstract
Solution phase ligand substitution reactions of [Pt(dien)Cl]+ and [Pt(Me5dien)Cl]+ (Me5dien = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) have been monitored using electrospray ionization mass spectrometry. The [Pt(Me5dien)Cl]+ ion undergoes much slower ligand substitution than [Pt(dien)Cl]+, which is consistent with the crystal structure of [Pt(Me5dien)Cl]+ determined, which suggests steric crowding (relative to the [Pt(dien)Cl]+ structure) induced by the buttressing methyl groups. In addition, the gas phase collision induced dissociation reactions of [Pt(dien)X]n+ ions (n = 1, X = bromide, iodide, thiocyanate, cyanate, azide or nitrite; n = 2, X = pyridine) formed via solution phase ligand substitution have been examined. In most cases the ligand is lost as HX, giving rise to the co-ordinatively unsaturated [Pt(dien) − H]+ ion. Noteable exceptions include n = 1, where X = N3− or NO2−, which instead exhibit ligand fragmentation via loss of N2 and NO respectively to yield the novel platinum species [Pt(dien)N]+ and [Pt(dien)O]+. The structures of these ions have been examined using density functional calculations at the B3LYP/LAV1S level of theory.
Introduction
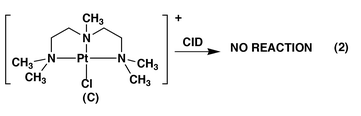
There has been considerable interest in exploiting electrospray ionization (ESI) mass spectrometry as a means of examining various aspects of metal ion chemistry, ranging from analytical issues (such as do the gas phase ion abundances in the mass spectrum reflect the solution phase metal ion speciation?)![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 1 to understanding the fundamental gas phase structure and reactivity of metal ions.2 Our interest has been in the latter area, where we have been examining: (i) the collision induced dissociation (CID) fragmentation reaction mechanisms of [Pt(terpy)M]2+ and [Pt(terpy)M − H]+ ions (where terpy = 2,2′∶6′,2′′-terpyridine; M = protein, nucleic acid or simple model system);3 (ii) the use of ion–molecule reactions as a means of probing co-ordinatively saturated versus co-ordinatively unsaturated platinum(II) ions.4 We found that CID of [Pt(dien)Cl]Cl A (dien = diethylenetriamine) results in HCl loss, eqn. (1), to yield the co-ordinatively unsaturated platinum ion B.4 Indirect evidence that the hydrogen atom lost was from one of the amino groups of the dien ligand comes from the CID spectrum of the related fully methylated system C, eqn. (2), which did not give facile HCl loss.
1 to understanding the fundamental gas phase structure and reactivity of metal ions.2 Our interest has been in the latter area, where we have been examining: (i) the collision induced dissociation (CID) fragmentation reaction mechanisms of [Pt(terpy)M]2+ and [Pt(terpy)M − H]+ ions (where terpy = 2,2′∶6′,2′′-terpyridine; M = protein, nucleic acid or simple model system);3 (ii) the use of ion–molecule reactions as a means of probing co-ordinatively saturated versus co-ordinatively unsaturated platinum(II) ions.4 We found that CID of [Pt(dien)Cl]Cl A (dien = diethylenetriamine) results in HCl loss, eqn. (1), to yield the co-ordinatively unsaturated platinum ion B.4 Indirect evidence that the hydrogen atom lost was from one of the amino groups of the dien ligand comes from the CID spectrum of the related fully methylated system C, eqn. (2), which did not give facile HCl loss.
| |

|
(1)
|
| |
|
(2)
|
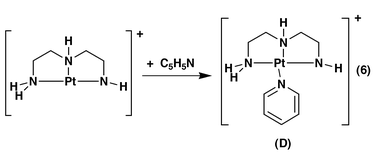
In addition, we found that complexes A and B exhibit completely different reactivity in gas phase ion–molecule reactions with neutral reagents such as water, methanol, acetonitrile and pyridine. For example, under our reaction conditions, A is unreactive towards pyridine in the gas phase, undergoing neither ligand substitution to give D, eqn. (3), deprotonation, eqn. (4), or formation of a hydrogen bonded adduct E, eqn. (5). In contrast, B reacts with Lewis bases such as pyridine to give the adduct D, eqn. (6).
| |

|
(3)
|
| |
| (6) |
Given that we have previously shown that complex A is unreactive towards ligand substitution, we herein address the following questions: (i) can we form new gas phase [Pt(dien)X]+ and [Pt(Me5dien)X]+ ions (Me5dien = pentamethyldiethylenetriamine) by carrying out ligand substitution reactions of A and B in the solution phase instead,5 and can we use ESI/MS to monitor these reactions; (ii) do gas phase [Pt(dien)X]+ ions fragment under CID conditions via HX loss to form B or can other novel platinum containing ions be formed? In this paper we also present details on the synthesis and characterization by single crystal X-ray diffraction of the platinum complex [Pt(Me5dien)Cl]+. In addition, density functional calculations have been carried out on various [Pt(dien)X]+ ions and fragment neutrals and ions in order to gain insights into the gas phase CID processes.
Experimental
Materials
The complexes [Pt(dien)Cl]Cl and cis-[Pt(dmso)2Cl2] were prepared according to the reported procedures.6,7
Chloro(N,N,N′,N′′,N′′-pentamethyldiethylenetriamine)platinum(II) perchlorate [Pt(Me5dien)Cl][ClO4]..
A solution of N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (0.25 g, 1.42 mmol) in methanol (20 ml) was added with stirring to a suspension of cis-[Pt(dmso)2Cl2] in methanol (100 mL). The mixture was heated at reflux for 1 h to form a pale yellow solution. The solvent was removed and the residue dissolved in water (15 mL). A saturated aqueous solution of NaClO4 (15 mL) was then added whereupon a pale yellow solid precipitated. The solid was redissolved by warming the mixture. On cooling overnight the product separated as pale yellow crystals which were collected and dried under high vacuum (0.54 g, 76%). Found: C, 21.65; H, 4.78; Cl, 14.13; N, 8.35. C9H23Cl2N3O4Pt requires C, 21.48; H, 4.61; Cl, 14.09; N, 8.35%. 195Pt NMR (D2O): δ
−2590.
Crystallography
A colourless plate approximately 0.36 × 0.34 × 0.06 mm was selected from crystals obtained from water.
Crystal data..
C9H23Cl2N3O4Pt, M = 503.29, tetragonal, space group I4/m (no. 87), a = 13.125(2), c = 19.049(3) Å, V = 3281.5(9) Å3, F(000) = 1936, Z = 8, Dc = 2.037 g cm−3, μ(Mo-Kα) = 8.89 mm−1, T = 293(1) K, Enraf-Nonius CAD-4MachS diffractometer, ω–2θ scans, 4582/1842 measured/unique data, Rint = 0.039. Absorption corrections applied.8
Structure determination..
The structure was solved by a combination of Patterson, direct methods and difference synthesis.9,10 All hydrogen atoms were included at geometrical estimates. The platinum, co-ordinated chlorine, and the central nitrogen of the triamine and its associated methyl carbon all lie on a mirror plane, giving 8 cations per unit cell. Four of the perchlorates lie on positions of 4 symmetry, while the other four are situated such that the chlorine and one oxygen lie on fourfold axes. A number of residual electron density peaks around both perchlorates indicated the anions were disordered: attempts to model this were unsuccessful. The perchlorates were constrained to tetrahedral geometry with a Cl–O distance of 1.44 Å. Anisotropic displacement factors were applied to each non-hydrogen atom, isotropic factors were used for hydrogens. Refinement against F2 using all data10 gave final R1 [1632 data, I = 2σ(I)] 0.0356, wR2 (all data) 0.0994. Neutral atom scattering factors and anomalous scattering correction terms were taken from ref. 11. Fig. 1 was prepared from the output of ORTEP II.12
![An ORTEP diagram of the [Pt(Me5dien)Cl]+ cation showing the labelling scheme; ellipsoids are at the 35% probability level.](/image/article/2000/DT/a908536j/a908536j-f1.gif) |
| | Fig. 1 An ORTEP diagram of the [Pt(Me5dien)Cl]+ cation showing the labelling scheme; ellipsoids are at the 35% probability level.
| |
CCDC reference number 186/1724.
See http://www.rsc.org/suppdata/dt/a9/a908536j/ for crystallographic files in .cif format.
Mass spectrometry
All experiments were performed using a commercially available quadrupole ion trap mass spectrometer (Finnigan-MAT model LCQ, San Jose, CA) equipped with electrospray ionization (ESI). For the experiments described, 71 μmol of inorganic salts or pyridine of reagent grade [specifically: potassium bromide (KBr) (Aldrich), sodium iodide (NaI) (Aldrich), potassium thiocyanate (KSCN) (Merck), sodium cyanate (NaOCN) (Merck), sodium azide (NaN3) (Ajax Laboratory Chemicals), sodium nitrite (NaNO2) (Ajax Laboratory Chemicals) and pyridine (C5H5N) (Ajax Laboratory Chemicals)] were each added to either [Pt(dien)Cl]Cl or [Pt(Me5dien)Cl][ClO4] in a 1∶1 ratio and dissolved in 1 mL of 1∶1 CH3CN–water. The 14 samples were then incubated at room temperature (298 K), and their ESI/MS recorded at intervals of t = 4, 12 and 24 h.
Typically, samples were further diluted ((0.1 mg mL−1) in 1∶1 CH3CN–water) prior to being introduced to the mass spectrometer at a flow rate of 3.0 μL min−1via electrospray ionization. The ESI conditions were: spray voltage, 5.0 kV; capillary temperature, 200 °C; nitrogen sheath gas pressure, 30 psi; and auxiliary gas flow rates, 20 (arbitrary units). The capillary voltage and tube lens offset voltage were both maintained at 0 V.
The three major isotopic ions from the [Pt(dien)X] or [Pt(Me5dien)X] isotopic clusters were mass selected (with a 5 m/z window) and subjected to CID to investigate their fragmentation reactions. Typically CID conditions were: activation amplitude, 0.35–0.60 V; activation (Q), 0.25 V; and activation time 100 ms.
Density functional theory calculations
Structures of ions and neutrals were optimized using density functional theory at the B3LYP level using the Jaguar13 program running on a Digital workstation. Given the “large size” of several of the systems calculated, the modest LAV1S basis set was used throughout.14 All optimized structures were then subjected to frequency calculations with the same basis set. Complete structural details and lists of vibrational frequencies for each of the optimized structures are available from the authors upon request.
Results and discussion
(A) Crystal structure of [Pt(Me5dien)Cl][ClO4]
An ORTEP diagram of the [Pt(Me5dien)Cl]+ cation is shown in Fig. 1, while selected bond lengths and bond angles are listed in Table 1. The Pt atom is in a square planar environment, co-ordinated to the three nitrogens of the ligand and a chloride ion. The environment is tetrahedrally distorted, with the Pt atom lying 0.066(4) Å below the least squares planes of the four donor atoms; N1 and N1′ (where ′ refers to that atom related by the symmetry operator x, y, −z ) lie above the Pt atom, while N2 and the Cl are situated below the Pt atom. This is in contrast to the pyramidal distortion reported for [Pt(Me5dien)(OMeC(O)NH2)]+15 but is similar to the tetrahedral distortion found for [Pt(dien)Cl]+, these minor distortions having been ascribed to packing effects.16 A comparison of our data in Table 1 with the data reported in refs. 15 and 16 reveals that the bond lengths and angles of the three complexes are similar. In particular, the bonds between the Pt atom and the terminal amines are significantly longer than the bond with the central amine, while the N1–Pt–N2 angles are significantly less than 90° and the N1–Pt–N1′ and N2–Pt–Cl angles are less than 180°. The main difference between the dien and the Me5dien complexes is that in the latter the angles around the nitrogen atoms are smaller due to the presence of the more sterically demanding methyl groups, which leads to a slight change in the conformation of the dien moiety.
Table 1 Selected bond lengths [Å] and angles [°] for [Pt(Me5dien)Cl]ClO4
| Symmetry transformation used to generate equivalent atoms: ′x,y,−z. |
| Pt–N2 |
2.013(8) |
Pt–N1 |
2.067(6) |
| Pt–Cl1 |
2.302(3) |
N1–C1 |
1.483(11) |
| N1–C2 |
1.493(10) |
N1–C3 |
1.497(11) |
| C3–C4 |
1.504(12) |
C4–N2 |
1.496(9) |
| N2–C5 |
1.509(13) |
|
|
| |
|
|
|
| N2–Pt–N1 |
85.96(18) |
N1′–Pt–N1 |
167.2(3) |
| N2–Pt–Cl1 |
177.6(2) |
N1–Pt–Cl1 |
94.25(18) |
| C1–N1–C2 |
108.8(7) |
C1–N1–C3 |
110.2(6) |
| C2–N1–C3 |
108.6(7) |
C1–N1–Pt |
108.1(5) |
| C2–N1–Pt |
114.3(5) |
C3–N1–Pt |
106.8(5) |
| N1–C3–C4 |
109.6(7) |
N2–C4–C3 |
108.1(6) |
| C4′–N2–C4 |
113.9(8) |
C4–N2–C5 |
110.0(5) |
| C4–N2–Pt |
105.0(5) |
C5–N2–Pt |
112.8(7) |
(B) Monitoring ligand substitution of [Pt(dien)Cl]+ and [Pt(Me5dien)Cl]+ by anionic inorganic nucleophiles and pyridine using ESI/MS
We have recently been examining the use of ESI-MS/MS to characterize the binding of platinum complexes of tridentate ligands such as terpyridine to biomolecules including amino acids and peptides.3 During the course of these studies we have observed that the formation of the platinum–biomolecule complexes is time dependent. We propose that the relative rates of substitution are strongly dependent on two factors: (a) the structure of the tridentate nitrogen ligands co-ordinated to the platinum atom and (b) the types of nucleophilic sites available for substitution on the amino acid or peptide. There are generally three or more different nucleophilic sites on any one amino acid or peptide and this indicated the need to study the ligand substitution reactions of simpler nucleophiles. Therefore, we became interested in monitoring the relative rates of substitution of the chlorine ligand of [Pt(dien)Cl]+, eqn. (7), and
[Pt(Me5dien)Cl]+, eqn. (8), by the simple anionic nucleophiles (X−) bromide, iodide, thiocyanate, cyanate, azide, nitrite as well as by the neutral nucleophile pyridine.
| | |
[Pt(dien)Cl]+ + X− → [Pt(dien)X]+ + Cl−
|
(7)
|
| | |
[Pt(Me5dien)Cl]+ + X− → [Pt(Me5dien)X]+ + Cl−
|
(8)
|
Thus the reactivity of [Pt(dien)Cl]+ and [Pt(Me5dien)Cl]+ towards nucleophilic displacement of the chloride ligand by X to produce [Pt(dien)X]+ and [Pt(Me5dien)X]+ ions was monitored over time by ESI/MS (Table 2, Fig. 2). After a period of 4 h significant differences in the ESI/MS spectra of ([Pt(dien)Cl]+ + X−) and ([Pt(Me5dien)Cl]+ + X−) were observed (Table 2). Generally, [Pt(dien)X]+ was the major ion observed in the ESI/MS spectra for ([Pt(dien)Cl]+ + X−), whereas the initial [Pt(Me5dien)Cl]+ ion was always the dominant ion observed in the ([Pt(Me5dien)Cl]+ + X−) ESI/MS spectra, even after a period of 24 h (Table 2). This result suggests the methyl groups of the [Pt(Me5dien)Cl]+ ion sterically hinder the access of even small nucleophiles to the platinum center, which is entirely consistent with the crystal structure described in section (A).
Table 2 Reactivity of platinum derivatives at t = 4, 12 and 24 h incubation time (T = 298 K)
| |
4 h |
12 h |
24 h |
| X− |
R = H |
R = CH3 |
R = H |
R = CH3 |
R = H |
R = CH3 |
|
At longer times the ESI spectra become more complicated, with ions of higher mass appearing.
Abundance of ion is less than 1%.
|
| Br− |
(B) major |
(A) major |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
| |
(A)b |
(B)b |
|
|
|
|
| I− |
(B) major |
(A) major |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
| |
(A)b |
(B)b |
|
|
|
|
| NO2− |
(B) major |
(A) major |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
| |
no (A) |
(B)b |
|
|
|
|
| OCN− |
(B) major |
no (B) |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
| |
(A)b |
(A) major |
|
|
|
|
| SCN− |
(B) major |
(A) major |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
| |
(A)b |
No (B) |
|
|
|
|
| N3− |
(B) major |
(A) major |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
Same as 4 ha |
| |
No (A) |
No (B) |
|
|
|
|
| C5H5N |
(B) major |
(A) major |
(B) major |
Same as 4 ha |
Same as 12 ha |
Same as 4 ha |
| |
(A)b |
(B)b |
No (A) |
|
|
|
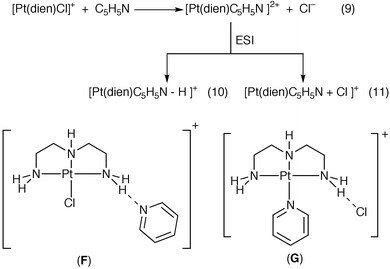
So what products would be observed by ESI/MS by reaction of these platinum complexes with a larger neutral nucleophile such as pyridine? In a similar fashion to the behaviour observed for the anionic nucleophiles, the [Pt(Me5dien)Cl] + ion is relatively unreactive towards pyridine (Fig. 2b). In contrast, three pyridine and platinum containing ions were observed in the ESI/MS spectrum of ([Pt(dien)Cl]Cl + pyridine) (Fig. 2a): the doubly charged [Pt(dien)(C5H5N)]2+, eqn. (9); the deprotonated singly charged [Pt(dien)(C5H5N) − H]+, eqn. (10); and the singly charged [Pt(dien)(C5H5N) + Cl]+ complex, eqn. (11). This latter ion may possess a number of different structures such as the ion–molecule complexes between [Pt(dien)Cl]+ and pyridine (i.e. structures such as E or F) or the ion pair complex17 between [Pt(dien)(C5H5N)]2+ and Cl− (i.e. structure G). In order further to investigate the structure of this complex, we have used CID as a probe, as described further in Section C.
(C) CID Reactions of [Pt(dien)X]+ (X = anionic ligand) under ESI-MS/MS conditions: competition between ligand loss and ligand fragmentation
Given that we are interested in using CID as both an analytical structural tool as well as a way of synthesizing novel platinum ions in the gas phase (for subsequent structure/reactivity studies using ion–molecule reactions), we sought to address some key issues regarding the CID process. In particular, for the CID fragmentation reactions of [PtII(L3)X]+ ions: (i) what role does the tridentate ligand L3 (where L3 is a tridentate ligand such as dien, Me5dien, terpy, etc.) have in controlling the fragmentation pathways available for the complex; (ii) what are the factors (e.g. bond strengths) which govern the competition between loss of the ligand X, e.g.eqn. (12) and fragmentation of the ligand X?18
| |
|
(9)
|
| |
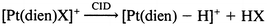
| (12) |
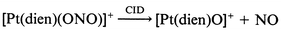
Our previous studies have shown that the tridentate ligand structure can have a profound influence on the fragmentation reaction channels available. Thus, the dien ligand has acidic N–H protons which facilitate ligand loss as the neutral species HX to yield ion B. Indeed, 5 of the 7 [Pt(dien)X]+ ions fragment exclusively via loss of HX (HX = HBr, HI, HOCN, HSCN or [C5H5NH]+), eqn. (12) and Fig. 3a. Note that the product of these reactions is the highly reactive, co-ordinatively unsaturated ion B, cf.eqn. (1).4 We have previously demonstrated that B can readily undergo gas phase ion–molecule reactions with adventitious background gases present in the ion trap (such as water) to give adduct ions (e.g. the ions at m/z 314, 315 and 316 in Fig. 3(a) are due to the water adduct).‡ Only two of the ligands were observed to undergo ligand fragmentation: thus the [Pt(dien)(NO2)]+ and [Pt(dien)(N3)]+ ions fragment to yield novel platinum ions, eqns. (13)–(15), Fig. 3(b), (c).
![ESI-MS/MS of [Pt(dien)X]+ ions: X = NCO− (a), N3− (b) or NO2− (c).](/image/article/2000/DT/a908536j/a908536j-f3.gif) |
| | Fig. 3 ESI-MS/MS of [Pt(dien)X]+ ions: X = NCO− (a), N3− (b) or NO2− (c).
| |
| |
|
(13)
|
| |
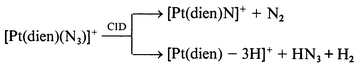
|
(14)
|
Can we observe any correlation between the types of CID products formed and gas phase thermochemistry? Unfortunately there is a dearth of thermochemical information on gas phase platinum complex ions. It seems reasonable to consider two accessible thermodynamic quantities: (i) the anion proton affinity of anionic ligand X−;19 (ii) bond strengths within the ligand X. Most of the nucleophiles which are lost as HX from the [Pt(dien)X]+ (Cl−, Br−, I− or SCN−) have proton affinities less than 1400 kJ mol−1 (the exception is OCN which has a proton affinity of 1443 kJ mol−1).19 The azide and nitrite ions which undergo ligand fragmentation have higher proton affinities (1418 and 1421 kJ mol−1 respectively), and might bind more strongly to Pt.19 Unfortunately, we cannot discuss the effects of the bond strengths in the various ligands as there are no data for the systems of interest co-ordinated to platinum.
In an attempt to gain some insights into the competition between ligand loss versus ligand fragmentation for the nitrite and azide systems, we have carried out density functional theory calculations at the B3LYP/LAV1S level of theory and our results are shown in Fig. 4 and Table 3. Note that this is a very modest level of theory and its reliability for predicting energies of both co-ordinatively saturated and unsaturated systems as well as “exotic platinum species” has yet to be established. Also, we have only calculated the reactants and products, but not the transition states for the competing fragmentation reactions (note that CID reactions are likely to be under kinetic rather than thermodynamic control). Not withstanding these cautionary notes, a number of interesting points emerge from these calculations. Regarding ligand loss with concomitant deprotonation at the dien ligand, two isomers for [Pt(dien) − H]+ were found. These correspond to deprotonation at the NH2 site (isomer 1, Fig. 4a) and the NH site (isomer 2, Fig. 4b). Isomer 1 shows a shortening of the Pt–N bond at the site of deprotonation and a lengthening of the trans Pt–N bond and is 49.5 kJ mol−1 less stable than isomer 2 (Table 3). Note that the kinetically favoured ligand loss channel may very well arise from deprotonation at the NH2 group since this is likely to have a lower transition state energy. This is because the NH2 protons are likely to be in closer geometric contact (e.g. see structure in Fig. 4e). An examination of the DFT results for the [Pt(dien)(N3)]+ system reveals the following points: (i) a bent structure is observed for the azide, as is expected for metal azides; (ii) the [Pt(dien)N]+ fragment ion has a considerably shortened Pt–N bond (Fig. 4g) consistent with multiple bonding and; (iii) the energetics of the two competing pathways (eqns. (12)) versus(14) indicates that the ligand fragmentation pathway resulting in formation of [Pt(dien)N]+ is thermodynamically favoured by over 20 kJ mol−1. Turning to our DFT results for the [Pt(dien)(ONO)]+ system we note the following: (i) two rotamers were found (Fig. 4d and 4e), with the one involving hydrogen bonding between the nitro oxygen and a H atom of the NH2 of the dien ligand being the most stable structure (Fig. 4e); (ii) the N bonded isomeric structure (Fig. 4f) is less stable by over 90 kJ mol−1; (iii) the [Pt(dien)O]+ fragment ion also has a considerably shortened Pt–O bond (Fig. 4h) consistent with multiple bonding, and (iv) the energetics of the two competing pathways (eqns. (12)versus(13) thermodynamically favours the ligand loss channel. This may indicate that the fragmentation reactions are under kinetic control, but further calculations at higher levels of theory may resolve this issue.
Table 3 Energies derived from DFT calculations (B3LYP/LAV1s)
| Structure |
B3LYP/LAV1sa |
ZPVEb |
Relative energyc/kJ mol−1 |
|
Absolute energies are in Hartrees.
ZPVE = Zero point vibrational energy, in kJ mol−1, taken from the frequency calculations.
Differences in energies are calculated using the absolute energies modified by the ZPVEs.
Isomer 1, structure shown in Fig. 4(a).
Isomer 2, structure shown in Fig. 4(b).
Structure shown in Fig. 4(c).
Rotamer 1, structure shown in Fig. 4(d).
Rotamer 2, structure shown in Fig. 4(e).
Structure shown in Fig. 4(f).
Structure shown in Fig. 4(g).
Structure shown in Fig. 4(h).
Product is isomer 1, Fig. 4(a).
Reactant is rotamer 2, Fig. 4(e).
|
| [Pt(dien) − H]+d |
−347.21481 |
492.2 |
0.0 |
| e |
−347.23386 |
492.8 |
−49.5 |
| [Pt(dien)(N3)]+f |
−509.86559 |
558.1 |
|
| [Pt(dien)(ONO)]+g |
−550.18629 |
554.0 |
54.5 |
| h |
−550.20765 |
555.6 |
0.0 |
| [Pt(dien)(NO2)]+i |
−550.17171 |
553.8 |
92.6 |
| [Pt(dien)N]+j |
−401.74647 |
537.7 |
| [Pt(dien)O]+k |
−421.82357 |
542.1 |
| HN3 |
−162.57514 |
52.6 |
| HONO (cis) |
−202.89928 |
53.2 |
| N2 |
−108.05604 |
13.4 |
| NO |
−128.13303 |
11.1 |
| Energetics (at the B3LYP/LAV1s + ZPE level) for reactions (12), (13) and (14) |
| Eqn. |
Energyc/kJ mol−1 |
Eqn. |
Energyc/kJ mol−1 |
| 12, X = NO2m |
235.6 |
13m |
656.8 |
| 12, X = N3l |
185.4 |
14 |
158.7 |
![B3LYP/LAV1S optimized structures of: (a) [Pt(dien) − H]+ isomer 1 (deprotonated at NH2 group); (b) [Pt(dien) − H]+ isomer 2 (deprotonated at NH) group; (c) [Pt(dien)(N3)]+; (d) [Pt(dien)(ONO)]+ rotamer 1 (no hydrogen bonding); (e) [Pt(dien)(ONO)]+ rotamer 2 (with hydrogen bonding); (f) [Pt(dien)(NO2)]+; (g) [Pt(dien)N]+; (h) [Pt(dien)O]+.](/image/article/2000/DT/a908536j/a908536j-f4.gif) |
| | Fig. 4 B3LYP/LAV1S optimized structures of: (a) [Pt(dien) − H]+ isomer 1 (deprotonated at NH2 group); (b) [Pt(dien) − H]+ isomer 2 (deprotonated at NH) group; (c) [Pt(dien)(N3)]+; (d) [Pt(dien)(ONO)]+ rotamer 1 (no hydrogen bonding); (e) [Pt(dien)(ONO)]+ rotamer 2 (with hydrogen bonding); (f) [Pt(dien)(NO2)]+; (g) [Pt(dien)N]+; (h) [Pt(dien)O]+.
| |
In order experimentally to examine whether ligand fragmentation (e.g. via loss of CO or CS for the cases of OCN and SCN) could be induced by “switching off” the competing HX ligand loss channel through the use of the Me5dien ligand, we have incubated [Pt(Me5dien)Cl]+ with AgNO3 in water (10 min at 100 °C, 6 h at 70 °C) to form [Pt(Me5dien)(OH2)]2+, which was then allowed to react with the appropriate nucleophile (OCN− and SCN−) at 70 °C for 12 h to generate the desired OCN− and SCN− complexes. Ligand fragmentations of the complexes via losses of CO and CS are not observed under CID conditions. Indeed, the CID fragmentation reactions of [Pt(Me5dien)X]+ (where X = N3−, NO2−, OCN− or SCN−) are rather complex and appear to involve the Me5dien ligand. Isotopic labelling studies are currently underway to unravel the mechanisms of these reactions.
Of the two ligand fragmentation channels observed for [Pt(dien)X]+, the loss of N2, eqn. (14) is the least surprising as this type of fragmentation is a common phenomenon for both inorganic and organic azides under energetic conditions (either thermal or photochemical).20 What is significant about the loss of N2 is that there appears to be no literature precedent for platinum(II) systems. The resultant ion is also interesting in terms of its electronic structure as it could exist in either the platinum(II) nitrene form H or as PtIV–N triple bond I. The low level DFT calculations (Fig. 4g) show a shortened Pt–N bond, suggesting structure I.
(D) CID Reactions of [Pt(dien)(C5H5N)]2+, [Pt(dien)(C5H5N) − H]+ and [Pt(dien)(C5H5N) + Cl]+ under ESI-MS/MS conditions
In order to examine the effect of the neutral ligand pyridine on the fragmentation reactions, the CID spectra of the [Pt(dien)(C5H5N)]2+ doubly charged (Fig. 5a) and the [Pt(dien)(C5H5N) − H]+ singly charged (Fig. 5b) ions were measured. Both ions fragment via ligand loss, resulting in the formation of ion B. In the case of the doubly charged ion the ligand is lost as protonated pyridine (m/z 80), which is also observed in the MS/MS spectrum (Fig. 5a). Presumably in this case neutral pyridine abstracts a proton from the tridentate dien ligand prior to fragmentation.
![ESI-MS/MS of (a) [Pt(dien)(C5H5N)]2+; (b) [Pt(dien)(C5H5N) − H]+ and (c) [Pt(dien)(C5H5N) + Cl]+.](/image/article/2000/DT/a908536j/a908536j-f5.gif) |
| | Fig. 5 ESI-MS/MS of (a) [Pt(dien)(C5H5N)]2+; (b) [Pt(dien)(C5H5N) − H]+ and (c) [Pt(dien)(C5H5N) + Cl]+.
| |
The structure of the [Pt(dien)(C5H5N) + Cl]+ ion was also probed via CID (Fig. 5c). We anticipate the type of neutral lost during the CID process will give some insight into the most stable gas phase structure of the [Pt(dien)(C5H5N) + Cl]+ ion (E, F, G). Thus, ion–molecule complexes between [Pt(dien)Cl]+ and pyridine (i.e. structures such as E or F) are likely to lose C5H5N, while the ion pair complex between [Pt(dien)(C5H5N)]2+ and Cl− (i.e. structure G) is likely to lose HCl via a deprotonation reaction.17 An examination of Fig. 3(c) reveals the exclusive loss of HCl to generate D, eqn. (16). This loss indicates that the ion pair complex structure G is the most important gas phase isomeric structure of the [Pt(dien)(C5H5N) + Cl]+ ion in the CID reaction. Unfortunately molecular modeling (even at the modest B3LYP/LAV1S level of theory) to probe the structures and relative stabilities of E, F and G was not possible with existing computational resources.
| |
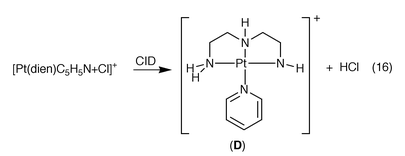
| (16) |
Conclusion
ESI/MS is a powerful tool to monitor solution phase ligand substitution reactions of platinum(II) complexes. A major strength of this technique is that when used in conjunction with tandem mass spectrometry it allows for the analysis of the substituted products. This aspect has been demonstrated in this work. Furthermore, from a fundamental aspect, it now becomes possible to “synthesize” novel platinum species which are co-ordinatively unsaturated or which have novel bonding arrangements from readily available co-ordinatively saturated precursors using CID. As a result, we are currently examining the use of gas phase ion–molecule reactions as structural probes of these novel CID products and the possibility of forming these species (e.g. [Pt(dien)N]+) in the condensed phase.
Acknowledgements
R. A. J. O. thanks the Australian Research Council for financial support and the University of Melbourne for funds to purchase the LCQ spectrometer. W. D. McF. thanks the NHMRC for financial support. M. L. S. acknowledges the award of a Commonwealth Postgraduate Scholarship.
References
-
Electrospray ionization mass spectrometry: fundamentals, instrumentation and applications, ed. R. B. Cole, Wiley, New York, 1997;
Search PubMed; R. Colton, A. D’Agostino and J. C. Traeger, Mass Spectrom. Rev., 1995, 14, 79 Search PubMed; J. W. Olesik, J. A. Kinser and S. V. Olesik, Spectrochim. Acta, Part B, 1998, 53, 239 CAS; G. R. Agnes, I. I. Stewart and G. Horlick, Appl. Spectrosc., 1994, 48, 1347 CrossRef.
- K. Eller and H. Schwarz, Chem. Rev., 1991, 91, 1121 CrossRef CAS;
Gas Phase Inorganic Chemistry, ed. D. H. Russell, Plenum, New York, 1989.
Search PubMed.
- R. A. J. O’Hair, Eur. Mass Spectrom., 1997, 3, 390 Search PubMed;
M. L. Styles
, R. A. J. O’Hair
and W. D. McFadyenmanuscript in preparation..
- G. E. Reid, R. A. J. O’Hair, M. L. S. Styles, W. D. McFadyen and R. J. Simpson, Rapid Commun. Mass Spectrom., 1998, 12, 1701 CrossRef CAS.
-
J. P. Collman
, L. S. Hegedus
, J. R. Norton
and R. G. Finke
, Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, CA, 1987, pp. 241–243;
Search PubMed; F. Basolo, H. B. Gray and R. G. Pearson, J. Am. Chem. Soc., 1960, 82, 4200 Search PubMed; H. B. Gray, J. Am. Chem. Soc., 1962, 84, 1548 CrossRef CAS.
- G. Annibale, M. Brandolisio and B. Pitteri, Polyhedron, 1995, 14, 451 CrossRef CAS.
- J. H. Price, A. S. Williamson, R. F. Schramm and B. B. Wayland, Inorg. Chem., 1972, 11, 1280 CrossRef CAS.
-
G. M. Sheldrick
, SHELX 76, Program for Crystal Structure Determination, University of Cambridge, 1976.
Search PubMed.
-
G. M. Sheldrick
, SHELXS 97, Program for Crystal Structure Solution, University of Göttingen, 1997.
Search PubMed.
-
G. M. Sheldrick
, SHELXL 97, Program for Crystal Structure Refinement, University of Göttingen, 1997.
Search PubMed.
-
International Tables for X-Ray Crystallography, Kluwer, Dordrecht, 1993, vol. C, p. 219.
Search PubMed.
-
C. K. Johnson
, ORTEP II, Fortran Thermal Ellipsoid Plot Program, Report ORNL-5138, Oak Ridge National Laboratories, Oak Ridge, TN, 1976.
Search PubMed.
-
Jaguar 3.0, Schrodinger, Inc., Portland, OR, 1997.
Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- R. Cini, F. P. Intini, L. Maresca, C. Pacifico and G. Natile, Eur. J. Inorg. Chem., 1998, 1305 CrossRef CAS.
- J. F. Britten, C. J. L. Lock and W. M. C. Pratt, Acta Crystallogr., Sect. B, 1982, 38, 2148 CrossRef.
- D. S. Gross and E. R. Williams, Int. J. Mass Spectrom. Ion Proc., 1996, 157/158, 305 CrossRef CAS.
- A. B. Norbury, Adv. Inorg. Chem. Radiochem., 1975, 17, 231 Search PubMed; J. L. Burmeister, R. L. Hassel, K. A. Johnson and J. C. Lim, Inorg. Chim. Acta, 1974, 9, 23 CrossRef CAS;
J. L. Burmeister
in Chemistry and Biochemistry of Thiocyanic Acid and its Derivatives, ed. A. A. Newman, Acadmic Press, London, 1975, ch. 2.
Search PubMed.
- Anion proton affinities are available from the NIST website http∶//webbook.nist.gov/chemistry..
- A. L. Poznyak and V. I. Pavlovski, Angew. Chem., Int. Ed. Engl., 1988, 27, 789 CrossRef.
Footnotes |
| † Supplementary data available: rotatable 3-D crystal structure diagram in CHIME format. See http://www.rsc.org/suppdata/dt/a9/a908536j/ |
| ‡ A reviewer has asked whether these ions are due to gas phase or solution phase processes. Since the precursor ions are mass selected prior to dissociation any other ions due to solution phase processes are ejected. Thus any new ions in the MS/MS spectrum must arise from CID processes or gas phase ion–molecule reactions. Indeed if B is mass selected (i.e. a MS3 experiment) and allowed to react with background gases for extended times (up to 10 s) these adduct ions appear. Furthermore, if water is deliberately introduced into the ion trap the ions at m/z 314, 315 and 316 dominate. |
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here. ![Electrospray ionisation ESI/MS of: (a) [Pt(dien)Cl]+ incubated with pyridine; (b) [Pt(Me5dien)Cl]+ incubated with pyridine. In both cases the incubation conditions were: molar ratio (1∶1), incubation time 24 h, solvent 50∶50 acetonitrile–water, room temperature (24 °C).](/image/article/2000/DT/a908536j/a908536j-f2.gif)
