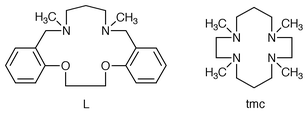
Kinetics and mechanisms of the oxidation of hypophosphite and phosphite with trans-[RuVI(L)(O)2]2+ (L = 1,12-dimethyl-3,4∶9,10-dibenzo-1,12-diaza-5,8-dioxacyclopentadecane)
Douglas T. Y.
Yiu
,
Kwok-Ho
Chow
and
Tai-Chu
Lau
*
Department of Biology and Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong, People’s Republic of China. E-mail: bhtclau@cityu.edu.hk
First published on 14th January 2000
Abstract
The kinetics of the oxidation of hypophosphite and phosphite by trans-[Ru(L)(O)2]2+ (L = 1,12-dimethyl-3,4∶9,10-dibenzo-1,12-diaza-5,8-dioxacyclopentadecane) have been studied in aqueous acidic solutions. The reactions have the following stoichiometry (x = 2 or 3): trans-[RuVI(L)(O)2]2+ + H2POx− + H2O → trans-[RuIV(L)(O)(OH2)]2+ + H2POx + 1−. The two reactions have the same rate law (P = hypophosphite or phosphite): −d[RuVI]/dt = k/(1 + [H+]/K)[RuVI][P]. For hypophosphite, k = (1.3 ± 0.1) dm3 mol−1 s−1 and K = (9.7 ± 0.5) × 10−2 mol dm−3 at 298 K and I = 1.0 mol dm−3. For phosphite, k = (4.8 ± 0.4) × 10−2 dm3 mol−1 s−1 and K = (1.2 ± 0.2) × 10−2 mol dm−3 at 298 K and I = 0.2 mol dm−3. The effects of temperature were studied from 15 °C to 40 °C. For hypophosphite, ΔH![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ‡ = (60 ± 2) kJ mol−1 and ΔS‡ = (−41 ± 4) J mol−1 K−1 at pH = 1.86 and I = 1.0 mol dm−3. For phosphite, ΔH‡ = (59 ± 4) kJ mol−1 and ΔS‡ = (−75 ± 13) J K−1 mol−1 at pH = 2.3 and I = 0.2 mol dm−3. Deuterium isotope effects have also been investigated. For hypophosphite, the kinetic isotope effect, k(H2PO2−)/k(D2PO2−) is 4.1 at pH = 1.07 and I = 1.0 mol dm−3. For phosphite, the kinetic isotopic effect, k(HDPO3−)/k(D2PO3−), is 4.0 at pH = 2.30 at I = 0.2 mol dm−3. A mechanism involving hydride transfer from P–H to Ru
‡ = (60 ± 2) kJ mol−1 and ΔS‡ = (−41 ± 4) J mol−1 K−1 at pH = 1.86 and I = 1.0 mol dm−3. For phosphite, ΔH‡ = (59 ± 4) kJ mol−1 and ΔS‡ = (−75 ± 13) J K−1 mol−1 at pH = 2.3 and I = 0.2 mol dm−3. Deuterium isotope effects have also been investigated. For hypophosphite, the kinetic isotope effect, k(H2PO2−)/k(D2PO2−) is 4.1 at pH = 1.07 and I = 1.0 mol dm−3. For phosphite, the kinetic isotopic effect, k(HDPO3−)/k(D2PO3−), is 4.0 at pH = 2.30 at I = 0.2 mol dm−3. A mechanism involving hydride transfer from P–H to Ru![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O is proposed for these two reactions.
O is proposed for these two reactions.
Introduction
Ruthenium forms an extensive series of oxo complexes with oxidation states ranging from IV to VIII.1 These oxo compounds are in general potent oxidants and there have been numerous reports on their reactions with organic substrates.1,2 We have been interested in studying the oxidation of inorganic substrates by these complexes, since much less is known in this area. These studies would also help us to understand the redox behaviour of ruthenium oxo complexes, many of which are potential oxidation catalysts. Che and co-workers have prepared a series of trans-dioxoruthenium(VI) complexes containing macrocyclic tertiary amine ligands,3–6 and their reactions with alkanes, alcohols and aromatic hydrocarbons have been studied.7–9 These complexes are particularly suitable for kinetic studies for the following reasons. The macrocyclic ligands are resistant to oxidative degradation and ligand exchange. These complexes also have well-defined redox potentials, and the reduced oxidation states RuV, RuIV and RuIII have been isolated and/or characterized spectroscopically. All these factors ensure clean kinetics, easy product identification and mechanistic interpretation. We have recently reported the kinetics and mechanisms of the oxidation of Fe2+(aq),10 iodide11 and sulfite12 by one of the macrocyclic dioxoruthenium(VI) complexes, trans-[RuVI(tmc)(O)2]2+ (tmc = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane). This complex is, however, only a mild oxidant; E° for the RuVI/V and the RuVI/IV couples are 0.56 V and 0.90 V (pH = 1.0) vs. NHE respectively;4 hence the range of substrates that can be oxidized by this complex is rather limited. We thus turn our attention to another dioxoruthenium(VI) complex, [Ru(L)(O)2]2+, which contains the macrocyclic ligand L (L = 1,12-dimethyl-3,4∶9,10-dibenzo-1,12-diaza-5,8-dioxacyclopentadecane). This complex is a much stronger oxidant, E° for the RuVI/V and the RuVI/IV couples are 0.94 V and 1.14 V (pH = 1.1) vs. NHE respectively.3 We report here kinetic studies of the oxidation of hypophosphite and phosphite by this complex in aqueous acidic solutions.
The predominant form of hypophosphorous acid is tetrahedral with two P–H bonds: H2P(O)OH. The predominant form of phosphorous acid is again tetrahedral but with only one P–H bond: HP(O)(OH)2.13 The protons in the P–H bonds are known to exchange only very slowly with D2O.14 The oxidation of these phosphorous oxoacids is of mechanistic interest since potential pathways may include one-electron oxidation, oxygen-atom transfer and hydride abstraction from P–H bonds. Although there are quite a number of reports on the oxidation of hypophosphite15–22 and phosphite,23–26 most of the kinetics were complicated by prior coordination of the phosphorous reductant to labile metal centres, or anhydride formation between a MO and a P–OH group. In the present case, prior coordination or anhydride formation may be excluded due to the presence of the non-labile and bulky macrocyclic ligand L. Thus less complicated kinetics are anticipated and more information on the redox process may be obtained.
Results and discussion
Spectral changes and stoichiometry
Preliminary repetitive scanning indicated rapid spectral changes when a solution of RuVI was mixed with a solution of hypophosphite (Fig. 1). The final spectrum showed quantitative formation of trans-[RuIV(L)(O)(OH2)]2+ (UV-Vis: λmax = 265 nm, ε = 2400 dm3 mol−1 s−1).3 Analysis by ion chromatography indicated that one mole of phosphite was produced from one mole of RuVI. Thus the reaction can be represented by the following equation:| [RuVI(L)(O)2]2+ + H2PO2− + H2O ⇌ [RuIV(L)O(OH2)]2+ + H2PO3− | (1) |
![Spectral changes during the oxidation of excess hypophosphite (1 × 10−3 mol dm−3) by trans-[RuVI(L)(O)2]2+ (1 × 10−4 mol dm−4) at T = 298.0 K, I = 1.0 mol dm−3 and pH = 1.9. Path length b = 1.0 cm. Time between consecutive runs = 60 s.](/image/article/2000/DT/a907664f/a907664f-f1.gif) | ||
| Fig. 1 Spectral changes during the oxidation of excess hypophosphite (1 × 10−3 mol dm−3) by trans-[RuVI(L)(O)2]2+ (1 × 10−4 mol dm−4) at T = 298.0 K, I = 1.0 mol dm−3 and pH = 1.9. Path length b = 1.0 cm. Time between consecutive runs = 60 s. | ||
Similar spectral changes were observed in the oxidation of phosphite and one mole of phosphate was formed from one mole of Ru(VI), indicating the following stoichiometry:
| [RuVI(L)(O)2]2+ + H2PO3− + H2O ⇌ [RuIV(L)O(OH2)]2+ + H2PO4− | (2) |
Kinetics
The kinetics for the oxidation of hypophosphite and phosphite were separately studied. Since the oxidation of hypophosphite by RuVI was found to be over 50 times faster than that of phosphite, there was no interference from the phosphite produced when hypophosphite was the substrate. The kinetic patterns for the oxidation of the two substrates were found to be the same, hence they are described together below. In the presence of at least a 10-fold excess of hypophosphite or phosphite, clean pseudo-first-order kinetics were observed for over three half-lives. The kinetics of the reactions were followed at 385 nm (λmax of RuVI). The pseudo-first-order rate constants, kobs, were independent of [RuVI] from 2.5 × 10−5 mol dm−3 to 1 × 10−4 mol dm−3. Thus the experimentally determined rate law is (P = hypophosphite or phosphite):| −d[RuVI]/dt = kobs[RuVI] = k2[RuVI][P] | (3) |
Representative results are summarized in Tables 1 and 2. The rate constant k2 was found to decrease with ionic strength, indicating a bimolecular reaction between ions of opposite charges. The effects of acid concentration on k2 were studied at 25.0 °C. The value of k2 was found to decrease with [H+], and a plot of 1/k2 against [H+] gave a straight line (Fig. 2 and 3). This is consistent with the following relationship:
| k 2 = k/(1+ [H+]/K) | (4) |
a for the oxidation of hypophosphite by [Ru(L)(O)2]2+
| T/K | I/mol dm−3 | [H+]/mol dm−3 | k 2/dm3 mol−1 s−1 |
|---|---|---|---|
| a pH was maintained with NaH2PO4 + CF3COOH. Ionic strength was adjusted with CF3CO2Na. b D3PO2 in D2O. c H3PO2 in D2O. | |||
| 298.0 | 1.0 | 3.23 × 10−1 | (3.05 ± 0.06) × 10−1 |
| 298.0 | 1.0 | 2.18 × 10−1 | (4.26 ± 0.09) × 10−1 |
| 298.0 | 1.0 | 1.12 × 10−1 | (6.25 ± 0.10) × 10−1 |
| 298.0 | 1.0 | 8.50 × 10−2 | (6.88 ± 0.10) × 10−1 |
| 298.0 | 1.0 | 8.50 × 10−2 | (1.11 ± 0.06) × 10−1b |
| 298.0 | 1.0 | 8.50 × 10−2 | (4.60 ± 0.10) × 10−1c |
| 298.0 | 1.0 | 5.25 × 10−2 | (8.54 ± 0.10) × 10−1 |
| 298.0 | 1.0 | 3.24 × 10−2 | 1.03 ± 0.03 |
| 298.0 | 1.0 | 1.38 × 10−2 | 1.14 ± 0.03 |
| 298.0 | 1.0 | 4.68 × 10−3 | 1.19 ± 0.03 |
| 298.0 | 1.0 | 1.55 × 10−3 | 1.48 ± 0.03 |
| 298.0 | 0.5 | 1.38 × 10−2 | 1.46 ± 0.03 |
| 298.0 | 0.2 | 1.38 × 10−2 | 2.17 ± 0.04 |
| 298.0 | 0.1 | 1.38 × 10−2 | 2.82 ± 0.06 |
| 288.0 | 1.0 | 1.38 × 10−2 | (4.47 ± 0.10) × 10−1 |
| 293.0 | 1.0 | 1.38 × 10−2 | (7.20 ± 0.10) × 10−1 |
| 303.0 | 1.0 | 1.38 × 10−2 | 1.68 ± 0.04 |
| 308.0 | 1.0 | 1.38 × 10−2 | 2.41 ± 0.08 |
| 313.0 | 1.0 | 1.38 × 10−2 | 3.77 ± 0.05 |
a for the oxidation of phosphite by [Ru(L)O2]2+
| T/K | I/mol dm−3 | [H+]/mol dm−3 | k 2/dm3 mol−1 s−1 |
|---|---|---|---|
| a pH was maintained with NaH2PO4 + CF3COOH. Ionic strength was adjusted with NaO2CCF3. b H3PO3 in D2O. c D3PO3 in D2O. | |||
| 298.0 | 0.2 | 2.24 × 10−2 | (1.63 ± 0.05) × 10−2 |
| 298.0 | 0.2 | 1.38 × 10−2 | (2.28 ± 0.10) × 10−2 |
| 298.0 | 0.2 | 1.11 × 10−2 | (2.51 ± 0.10) × 10−2 |
| 298.0 | 0.2 | 5.01 × 10−3 | (3.16 ± 0.12) × 10−2 |
| 298.0 | 0.2 | 5.01 × 10−3 | (1.90 ± 0.08) × 10−2b |
| 298.0 | 0.2 | 5.01 × 10−3 | (4.72 ± 0.19) × 10−3c |
| 298.0 | 0.2 | 1.70 × 10−3 | (4.35 ± 0.16) × 10−2 |
| 298.0 | 1.0 | 5.01 × 10−3 | (1.93 ± 0.08) × 10−2 |
| 293.0 | 0.2 | 5.01 × 10−3 | (2.01 ± 0.08) × 10−2 |
| 303.0 | 0.2 | 5.01 × 10−3 | (4.32 ± 0.16) × 10−2 |
| 308.0 | 0.2 | 5.01 × 10−3 | (5.96 ± 0.23) × 10−2 |
| 313.0 | 0.2 | 5.01 × 10−3 | (1.13 ± 0.05) × 10−1 |
![Plot of 1/k2vs. [H+] for the oxidation of hypophosphite by trans-[RuVI(L)(O)2]2+ at T = 298.0 K and I = 1.0 mol dm−3.](/image/article/2000/DT/a907664f/a907664f-f2.gif) | ||
| Fig. 2 Plot of 1/k2vs. [H+] for the oxidation of hypophosphite by trans-[RuVI(L)(O)2]2+ at T = 298.0 K and I = 1.0 mol dm−3. | ||
![Plot of 1/k2vs. [H+] for the oxidation of phosphite by [RuVI(L)(O)2]2+ at T = 298.0 K and I = 0.2 mol dm−3.](/image/article/2000/DT/a907664f/a907664f-f3.gif) | ||
| Fig. 3 Plot of 1/k2vs. [H+] for the oxidation of phosphite by [RuVI(L)(O)2]2+ at T = 298.0 K and I = 0.2 mol dm−3. | ||
Values of k and K were obtained from linear least-squares fits of the data to eqn. (4). For hypophosphite, k = (1.3 ± 0.1) dm3 mol−1 s−1 and K = (9.7 ± 0.5) × 10−2 mol dm−3 at 298 K and I = 1.0 mol dm−3. For phosphite, k = (4.8 ± 0.4) × 10−2 dm3 mol−1 s−1 and K = (1.2 ± 0.2) × 10−2 mol dm−3 at 298 K and I = 0.2 mol dm−3.
The effects of temperature were studied from 15 °C to 40 °C. Activation parameters were obtained from plots of ln (k2/T) versus 1/T according to the Eyring equation. For hypophosphite, ΔH‡ = (60 ± 2) kJ mol−1 and ΔS‡ = (−41 ± 4) J mol−1 K−1 at pH = 1.86 and I = 1.0 mol dm−3. For phosphite, ΔH‡ = (59 ± 4) kJ mol−1 and ΔS‡ = (−75 ± 13) J K−1 mol−1 at pH = 2.3 and I = 0.2 mol dm−3.
Deuterium isotope effects have also been investigated. For hypophosphite, the kinetic isotope effect, k(H2PO2−)/k(D2PO2−) is 4.1 at pH = 1.07 and I = 1.0 mol dm−3. For phosphite, the kinetic isotopic effect, k(HDPO3−)/k(D2PO3−), is 4.0 at pH = 2.30 and I = 0.2 mol dm−3.
Mechanism
The observed acid dependence of the rate constants for the two substrates is consistent with the anion H2POx− (x = 2 or 3) being the active species. The large kinetic isotopic effects indicate that the activation process involves P–H bond breaking. Since the ruthenium oxidant is already coordinated to a bulky macrocyclic ligand, coordination of the phosphorous substrate to the metal centre prior to the redox process can be excluded. Thus the most likely pathway that accounts for the large isotope effect involves direct P–H hydride or hydrogen-atom abstraction by a RuO bond. Although kinetically it is often difficult to distinguish a two-electron hydride abstraction pathway from a one-electron hydrogen-atom abstraction followed by another rapid one-electron transfer, the hydride abstraction pathway (eqn. (5)) is preferred for the following reasons. Intermediate formation of RuV was not observed even on a stopped-flow spectrophotometer. Moreover, a two-electron change is thermodynamically more favourable than a one-electron change for both the RuVI oxidant and the phosphorous reductants. At pH < 5, the RuVI/IV couple is more positive than the RuVI/V couple.3 Also a one-electron oxidation of PI or PIII would generate an unstable radical intermediate.
The proposed mechanism for the oxidation of hypophosphite and phosphite by trans-[RuVI(L)(O)2]2+ is shown in eqn. (5)–(7) below:
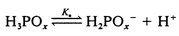
| (5) |
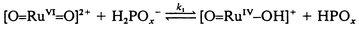
| (6) |
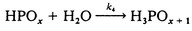
| (7) |
This mechanism leads to the observed rate law with k = k1 and K = Ka under the condition that the k4 step competes efficiently with k−1. The experimental value of Ka for hypophosphorous acid (9.7 × 10−2 mol dm−3) is in reasonable agreement with literature values of 1.07 × 10−1 mol dm−311 and 5.7 × 10−2 mol dm−3 (at infinite dilution).27 For phosphorous acid, the experimental Ka value (1.2 × 10−2) is quite different from the literature value of 5.5 × 10−2 mol dm−3 at infinite dilution.26 However, it is known from previous studies that the acidities of hypophosphorous acid and phosphorous acid are rather dependent on the ionic strength, the other species present and the temperature of the solution.
In the oxidation of hypophosphite by another metal-oxo species, [CrVIL′2(O)(OH)]− (L′ = 2-ethyl-2-hydroxybutanoate), a substrate isotopic effect of 3.9 was observed and a hydride abstraction mechanism was also proposed.21 In the oxidation of phosphite by HCrO4−, a large substrate isotope effect of ≈4 was also reported, but a mechanism involving initial anhydride formation and base removal of H+ by H2O and/or H2PO2− was proposed.13 It is possible that a hydride abstraction mechanism was also operating in this case. In this regard the oxidation of these phosphorous oxoacids by metal-oxo species is analogous to the oxidation of alcohols, where there is also strong evidence for a hydride abstraction mechanism.28,29
In conclusion, trans-[Ru(L)(O)2]2+ oxidizes hypophosphite and phosphite cleanly and efficiently, and most likely through a hydride abstraction mechanism, without any need for prior coordination of the phosphorous oxoacids to the metal centre. This is different from previous studies involving mostly labile metal oxidants, where there was strong evidence for prior coordination or anhydride formation.
Experimental
Materials
trans-[RuVI(L)(O)2][PF6]2 was prepared by a literature method3 (Found: C, 33.3; H, 3.9; N, 3.5. Calc. for C21H28N2O4P2F12Ru: C, 33.0; H, 3.7; N, 3.7%.) Hypophosphorous acid (50 wt% in water, RDH), hypophosphorous acid-d3 (50 wt% in D2O, 99+ atom% D, Aldrich) and phosphorous acid (30% in water, Merck) were used as received. They were standardized by titration with cerium(IV).30 D3PO4 was prepared by repeatedly dissolving H3PO4 in D2O, refluxing for 8 h, and evaporating excess D2O under vacuum.14 Ionic strength was maintained with sodium trifluoroacetate. Water for kinetic experiments was distilled twice from alkaline permanganate. The pH of solutions used for kinetic experiments was determined with a Corning Model 250 Ion Analyzer and a Hanna Instrument glass electrode after calibration with standard buffers at 25 °C. For D2O solutions pD was determined either by direct titration with standard NaOH solutions and/or by using the pH meter using the relationship pD = pHmeas + 0.4.
Kinetics
Kinetic experiments were performed under pseudo-first-order conditions using a Hewlett-Packard 8452A diode-array spectrophotometer. The progress of the reaction was monitored by measuring absorbance changes at 385 nm (λmax of RuVI). Pseudo-first-order rate constants, kobs, were obtained by nonlinear least-squares fits of At to time t according to the equation At = A∞ +(A0 − A∞)exp(−kobst), where A0 and A∞ are the initial and final absorbances, respectively.Products
In the oxidation of hypophosphite, analysis of phosphorus containing products was done by ion chromatography (IC) using a Wescan ICM 300 ion chromatograph equipped with an Alltech 335 suppressor module and an Alltech Allsep anion column. The mobile phase was 0.9 mmol dm−3 Na2CO3 and 0.9 mmol dm−3 NaHCO3. In a typical experiment a solution containing 1.4 × 10−4 mol dm−3 of [Ru(L)(O)2]2+ and a 8.0 × 10−4 mol dm−3 solution of hypophosphite in 1 × 10−3 mol dm−3 of CF3CO2H was prepared. After a reaction time of 2 h analysis of the solution by ion chromatography indicated the presence of 1.2 × 10−4 mol dm−3 of phosphite. Thus 1 mol of Ru(VI) produced 1 mol of phosphite.In the oxidation of phosphite, the product phosphate was analyzed by colorimetry using the phosphovanadomolybdate method.29 In a typical experiment a solution containing 8.0 × 10 −4 mol dm−3 of [Ru(L)(O)2]2+ and a 4.0 × 10−3 mol dm−3 solution of hypophosphite in 1 × 10−3 mol dm−3 of CF3CO2H was prepared. After a reaction time of 2 h, the solution was passed through a Sephadex SP C-25 cation exchange column. Vanadate-molybdate reagent was then added and the absorbance at 470 nm measured. 7.4 × 10−4 mol dm−3 of phosphate was detected, indicating that 1 mol of Ru(VI) produced 1 mol of phosphate.
Acknowledgements
Financial support from the Hong Kong Research Grants Council and the City University of Hong Kong is gratefully acknowledged.References
- C. M. Che and V. W. W. Yam, Adv. Inorg. Chem. Radiochem., 1992, 39, 233 Search PubMed.
- W. P. Griffith, Transition Met. Chem., 1990, 15, 251 Search PubMed; W. P. Griffith, Chem. Soc. Rev., 1992, 21, 179 RSC.
- C. M. Che, W. T. Tong, W. T. Wong and T. F. Lai, J. Am. Chem. Soc., 1989, 111, 9048 CrossRef CAS.
- C. M. Che, T. F. Lai and K. Y. Wong, Inorg. Chem., 1987, 22, 2289 CrossRef.
- C. M. Che, W. T. Tang, W. O. Lee, W. T. Wong and T. F. Lai, J. Chem. Soc., Dalton Trans., 1989, 2011 RSC.
- C. M. Che, W. T. Tang and C. K. Li, J. Chem. Soc., Dalton Trans., 1990, 3735 RSC.
- C. M. Che, W. T. Tang, W. O. Lee, K. Y. Wong and C. K. Li, J. Chem. Soc., Dalton Trans., 1992, 1551 RSC.
- C. M. Che, W. T. Tang, K. Y. Wong and C. K. Li, J. Chem. Soc., Dalton Trans., 1991, 3277 RSC.
- C. M. Che, C. K. Li, W. T. Tang and W. Y. Yu, J. Chem. Soc., Dalton Trans., 1992, 3153 RSC.
- T. C. Lau, K. W. C. Lau and C. K. Lo, Inorg. Chim. Acta, 1993, 209, 89 CrossRef CAS.
- T. C Lau, K. W. C. Lau and K. Lau, J. Chem. Soc., Dalton Trans., 1994, 3091 RSC.
- T. C. Lau, K. H. Chow, K. W. C. Lau and W. Y. K. Tsang, J. Chem. Soc., Dalton Trans., 1997, 313 RSC.
- N. N. Greenwood and A. Earnshaw , Chemistry of the Elements, Butterworth-Heinemann, Oxford, 2nd edn., 1997, p. 510. Search PubMed.
- A. Fratiello and E. W. Anderson, J. Am. Chem. Soc., 1963, 90, 519.
- G. P. Haight, Jr., M. Rose and J. Preer, J. Am. Chem. Soc., 1968, 90, 4809 CrossRef.
- J. N. Cooper, J. Phys. Chem., 1970, 74, 955 CrossRef CAS.
- K. S. Gupta and Y. K. Gupta, Inorg. Chem., 1974, 13, 851 CrossRef CAS.
- R. R. Nagori, M. Mehta and R. N. Mehrota, J. Chem., Soc., Dalton Trans., 1979, 216 RSC.
- A. K. Indrayan, S. K. Mishra and Y. K. Gupta, Inorg. Chem., 1981, 20, 1924 CrossRef CAS.
- S. K. Ghosh, R. N. Bose, K. Laali and E. S. Gould, Inorg. Chem., 1986, 25, 4737 CrossRef CAS.
- R. N. Mehrota and L. J. Kirschenbaum, Inorg. Chem., 1989, 28, 4327 CrossRef.
- R. Banerjee, R. Das and S. Mukhopadhyay, J. Chem. Soc., Dalton Trans., 1992, 1317 RSC.
- S. K. Chandra, E. Gelerinter and E. S. Gould, Inorg. Chem., 1995, 34, 4507.
- S. K. Mishra, P. D. Sharma and Y. K. Gupta, J. Inorg. Nucl. Chem., 1974, 36, 1845 Search PubMed.
- K. S. Gupta and Y. K. Gupta, J. Chem. Soc. A, 1971, 1180 RSC.
- R. N. Mehrotra, J. Chem. Soc., Dalton Trans., 1984, 1531 RSC.
- J. W. Larson and M. Pippin, Polyhedron, 1989, 8, 527 CrossRef CAS.
- S. L. Scott, A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1992, 114, 4205 CrossRef CAS.
- L. Roecker and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 746 CrossRef CAS.
- G. H. Jeffery , J. Basset , J. Mendham and R. C. Denney , Vogel’s Textbook of Quantitative Chemical Analysis, Longman, Essex, 5th edn., 1989. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |