Dinuclear and tetranuclear copper(II) complexes with bridging (N–N) diazine ligands: variable magnetic exchange topologies![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) †
†
Received
(in Cambridge, UK)
13th September 1999
, Accepted 10th November 1999
First published on 14th January 2000
Abstract
Dinuclear and tetranuclear copper(II) complexes of three polydentate diazine ligands (pahap, pmhap, bdpah), based on potentially bridging N–N single bond fragments, are reported. The 2∶2 complexes [Cu2(pahap)2(NO3)(H2O)2][NO3]3·H2O (1) and [Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2) involve dinuclear centres bridged by two N–N single bonds, and are essentially uncoupled, as a result of strict orbital orthogonality or an acute twisting of the copper magnetic planes relative to the N–N bridge. A mixed, double bridged (pyridazine/N–N) complex [Cu2(pahap)(dppn)(NO3)(H2O)][NO3]3 (3) has a larger angle between the magnetic planes, resulting in weak antiferromagnetic behaviour (2J = −32.5 cm−1). The rotationally more flexible 1∶1 complexes [Cu2(pahap)(C2O4)2]·0.5H2O (4) (2J = −4.4 cm−1) and [Cu2(pahap)(acac)2(H2O)2][NO3]2·H2O (5) (2J = −69.7 cm−1) involve single N–N bridges, and the angle between the copper magnetic planes depends on steric factors and hydrogen bonding interactions respectively. A tetranuclear complex [Cu2(pahap − H)(dpa)]2[NO3]4·4H2O (6) results from the linking of two {Cu2(N–N)} subunits with a secondary ligand strap, 1,3-diamino-2-propanol, and leads to a trans Cu2(N–N) bridging arrangement and strong antiferromagnetic coupling (2J = −186.4 cm−1). The complex [Cu2(bdpah − H)(NO3)2][NO3] (7) has a locked conformation with a large Cu–N–N–Cu torsional angle (130.3°), leading to moderately strong antiferromagnetic coupling (2J = −112.0 cm−1). The magnetic results are entirely consistent with previous magnetostructural correlations relating the twist of the copper magnetic planes around the N–N bond to the exchange integral.
Introduction
N2 diazine bridges found in some conjugated aromatic heterocyclic ligands can bring two copper(II) centers into close proximity and generate moderate to strong antiferromagnetic intramolecular exchange between the two copper centers via the p orbital system (σ pathway) of the heterocyclic ligand. This varies with the nature of the diazine ligand. Extensive studies have revealed that for dinuclear copper(II) complexes containing two essentially planar bis-(N4) ligands in which the magnetic orbital is dx2![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) −y2, the −2J values are in the order pyridazine/phthalazine (450–550 cm−1)1–4 > pyrazolate (200–430 cm−1)5–7 > triazolate (200–240 cm−1)2,8 > 4-aminotriazole (<220 cm−1).9,10 Compared with the diazine moiety in heterocyclic ring systems the N2 diazine linkages in open-chain systems with N–N single bonds are much more flexible.11 Previous studies12–16 showed that such open-chain diazine ligands present several possible mononucleating and dinucleating coordination modes due to the flexibility of the ligand around the N–N single bond. In combination with other donors such open–chain diazine ligands can form several types of copper(II) complex, e.g. mononuclear,16–19 dinuclear,20–26 trinuclear27 and tetranuclear.28,29 In our recent papers30,31 a series of novel open-chain diazine ligands has been shown to coordinate to two copper(II) centers as N4 or N5 donors with a variety of geometrical arrangements, which depend on co-ligands and reaction conditions. These flexible geometrical arrangements result from the ability of the systems to rotate freely about the single N–N bond of the diazine fragment. A linear relationship has been found between the rotation angle and the exchange integral over a 105° range for such dicopper(II) systems. When the rotation angle is less than 80° ferromagnetic exchange prevails.30,31
−y2, the −2J values are in the order pyridazine/phthalazine (450–550 cm−1)1–4 > pyrazolate (200–430 cm−1)5–7 > triazolate (200–240 cm−1)2,8 > 4-aminotriazole (<220 cm−1).9,10 Compared with the diazine moiety in heterocyclic ring systems the N2 diazine linkages in open-chain systems with N–N single bonds are much more flexible.11 Previous studies12–16 showed that such open-chain diazine ligands present several possible mononucleating and dinucleating coordination modes due to the flexibility of the ligand around the N–N single bond. In combination with other donors such open–chain diazine ligands can form several types of copper(II) complex, e.g. mononuclear,16–19 dinuclear,20–26 trinuclear27 and tetranuclear.28,29 In our recent papers30,31 a series of novel open-chain diazine ligands has been shown to coordinate to two copper(II) centers as N4 or N5 donors with a variety of geometrical arrangements, which depend on co-ligands and reaction conditions. These flexible geometrical arrangements result from the ability of the systems to rotate freely about the single N–N bond of the diazine fragment. A linear relationship has been found between the rotation angle and the exchange integral over a 105° range for such dicopper(II) systems. When the rotation angle is less than 80° ferromagnetic exchange prevails.30,31
The present study describes a series of dinuclear and tetranuclear copper(II) complexes, in which the copper centres are bridged by single N–N bonds from the polydentate ligands pahap, pmhap and bdpah (Scheme 1). The 2∶2 dinuclear complexes [Cu2(pahap)2(NO3)(H2O)2][NO3]3·H2O (1), [Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2) and [Cu2(pahap)(dppn)(NO3)(H2O)][NO3]3 (3) (dppn = 3,6-bis-(2′-pyridyl)pyridazine) differ from the previous examples in that the two metals are bridged by two N–N groups, and as a result are conformationally locked with acute angles between the copper magnetic planes, resulting in insignificant coupling or weak antiferromagnetic exchange between copper(II) centres (−2J < 35 cm−1). More rotational freedom exists in the dinuclear compounds [Cu2(pahap)(C2O4)2]·0.5H2O (4) and [Cu2(pahap)(acac)2(H2O)2][NO3]2·H2O (5), with rotational angles between the copper magnetic planes that are controlled by steric factors (4) and hydrogen bonding interactions (5). This leads to weak antiferromagnetic coupling for 4 (2J = −4.4 cm−1) and stronger antiferromagnetic coupling for 5 (2J = −69.7 cm−1). The tetranuclear complex [Cu2(pahap − H)(dpa)]2[NO3]4·4H2O (6) (Hdpa = 1,3-diamino-2-propanol) involves two dinuclear halves with a trans N–N bridging arrangement, and the two halves linked by orthogonal alkoxide bridges. This leads to strong antiferromagnetic exchange (2J = −186.4 cm−1). The conformationally locked complex [Cu2(bdpah − H)(NO3)2][NO3] (7) has a large rotational angle of the copper magnetic planes about the N–N bond (Cu–N–N–Cu torsional angle 130.3°), leading to moderately strong antiferromagnetic exchange (2J = −112.0 cm−1).
Experimental
Materials
Commercially available solvents and chemicals were used without further purification.
Physical measurements
Melting points were measured on a Fisher–Johns melting point apparatus. Electronic spectra were recorded as Nujol mulls and in solution using a Cary 5E spectrometer. Infrared spectra were recorded as Nujol mulls using a Mattson Polaris FTIR instrument. Mass spectra were obtained using a VG micromass 7070HS spectrometer. C, H, N analyses on vacuum dried samples (24 h) were performed by the Canadian Microanalytical Service, Delta, B.C., Canada. Variable temperature magnetic data (4–300 K) were obtained using an Oxford Instruments Superconducting Faraday Susceptometer with a Sartorius 4432 microbalance (main solenoid field of 1.5 T and a gradient field of 10 T m−1), and with a Quantum Design MPMS5S Squid magnetometer operating at 0.2 T (3–300 K). Calibrations were carried out with HgCo(NCS)4 and a palladium standard cylinder, and temperature errors were determined with [H2tmen][CuCl4] (H2tmen=(CH3)2HNCH2CH2NH(CH3)22+).32
Ligand preparations
Pahap, and pmhap were prepared by procedures described in previous reports.30,31
Bdpah..
The methyl ester of imino-2-pyridinecarboxylic acid was prepared in situ by reaction of 2-cyanopyridine (5.70 g, 54.8 mmol) with sodium methoxide solution, produced by dissolving sodium metal (0.13 g, 5.5 mmol) in dry methanol (50 cm3). This solution was then treated with biacetyl hydrazone (3.00 g, 25.9 mmol), prepared from 2,3-butanedione and 85% hydrazine hydrate in aqueous ethanol (1∶1) according to a standard literature procedure,12a and the mixture was refluxed for 24 h. The yellow solution was reduced in volume (ca. 10 cm3) and added slowly to a large volume of water (500 cm3) with stirring followed by 2–3 drops of glacial acetic acid. A yellow precipitate separated after 2 h, which was filtered off, washed with water (100 cm3), ethanol (100 cm3), diethyl ether (100 cm3) and dried under vacuum. Yield (6.82 g, 82%), mp 215–217 °C (Found: C, 59.30; H, 5.60; N, 34.74. C16H18N8 requires C, 59.61; H, 5.63; N, 34.76%); νmax/cm−1 3476 (NH), 3430 (NH), 3364 (NH), 3311 (NH), 1613 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N) and 996 (py) (Nujol); δH(CDCl3) 2.51 (s, 6H, CH3), 6.18 (br s, 4H, NH2), 7.38 (ddd, 2H, H-5), 7.80 (dt, 2H, H-4), 8.42 (d, 2H, H-6) and 8.61 (dd, 2H, H-3); m/z 322 (M+).
N) and 996 (py) (Nujol); δH(CDCl3) 2.51 (s, 6H, CH3), 6.18 (br s, 4H, NH2), 7.38 (ddd, 2H, H-5), 7.80 (dt, 2H, H-4), 8.42 (d, 2H, H-6) and 8.61 (dd, 2H, H-3); m/z 322 (M+).
Complex preparations
[Cu2(pahap)2(NO3)(H2O)2][NO3]3·H2O (1)..
Method A..
0.24 g (1.0 mmol) of pahap was added to a solution of the complex [Cu2(pahap)(H2O)6][NO3]430 (0.72 g, 1.0 mmol dissolved in 20 cm3 deionized water), forming a clear deep green solution after a few minutes. The solution was filtered and allowed to stand at room temperature for a few days. Deep green crystals, suitable for structural analysis formed, which were filtered off and dried in air. (Yield 85%) (Found: C, 31.56; H, 3.34; N, 24.50. [Cu2(C12H12N6)2(NO3)(H2O)2][NO3]3·H2O: requires C, 31.69; H, 3.32; N, 24.64%). λmax/nm (Nujol) 705; νmax/cm−1 3510, 3525 (H2O), 3356 (NH), 1756 (ν1 + ν4 NO3−), 1665, 1644 (CN) and 1024, 1013 (py) (Nujol).
Method B..
This complex was also synthesized in identical yield by mixing equimolar amounts (0.48 g, 2.0 mmol) of pahap and copper(II) nitrate (0.46 g, 2.0 mmol) in 20 cm3 deionized water.
[Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2)..
This compound was prepared as green crystals in a similar manner (method A or B) to compound 1, using pmhap. (Yield 85%) (Found: C, 34.82; H, 3.46; N, 21.94. [Cu2(C13H13N5)2(NO3)2][NO3]2·3H2O requires C, 34.75; H, 3.48; N, 21.82%). λmax/nm (Nujol) 719; νmax/cm−1 3500 (H2O), 3340 (NH), 1763 and 1749 (ν1 + ν4 bidentate and ionic NO3−), 1664 (CN) and 1044, 1017 (py) (Nujol).
[Cu2(pahap)(dppn)(H2O)(NO3)][NO3]3 (3)..
Dppn33 (0.243 g; 1.00 mmol) was added to a solution of the complex [Cu2(pahap)(H2O)6][NO3]430 (0.72 g, 1.0 mmol) dissolved in 20 cm3 deionized water, forming a clear green solution immediately. The solution was filtered and allowed to stand at room temperature for a few days. Green crystals suitable for structural analysis formed. (Yield 85%) (Found: C, 35.80; H, 2.90; N, 22.52. [Cu2(C12H12N6)(C14H10N4)(NO3)(H2O)][NO3]3 requires C, 35.99; H, 2.79; N, 22.60%). λmax/nm (Nujol) 650; νmax/cm−1 3310 (NH), 1764,1774 and 1724 (ν1 + ν4 monodentate and ionic NO3−), 1686, 1662 (CN) and 1036, 1026 (py) (Nujol).
[Cu2(pahap)(C2O4)2]·0.5H2O (4)..
Aqueous solutions of [Cu2(pahap)(H2O)6][NO3]430 (0.34 g, 0.47 mmol) and K3[Cr(C2O4)3]·3H2O (0.23 g, 0.47 mmol) were slowly diffused into one another using an H-tube apparatus. Small emerald green crystals, suitable for X-ray structural determination formed over a period of 3 months; these were filtered off and air-dried. Yield (76%) (Found: C, 34.56; H, 2.37; N, 15.21. [Cu2(C12H12N6)(C2O4)2]·0.5H2O requires C, 34.79; H, 2.37; N, 15.21%); λmax/nm (Nujol) 653; νmax/cm−1 3570 (OH), 3444–3170 (NH), 1673 (CO), and 1029 (py) (Nujol). The same compound can be prepared as a green powder (yield 90%) by mixing aqueous solutions of [Cu2(pahap)(H2O)6][NO3]430 and Na2C2O4 (1∶2) directly.
[Cu2(pahap)(acac)2(H2O)2][NO3]2·H2O (5)..
Acetylacetone (0.20 g, 2.0 mmol) was added to 5 cm3 of an aqueous solution of 0.4 M KOH, and the resulting solution added slowly to a solution of [Cu2(pahap)(H2O)6][NO3]430 (0.72 g, 1.0 mmol) in water (10 cm3) with stirring. Dark blue crystals suitable for structural analysis, formed on standing over several days (yield 80%). These were filtered off, washed with a small amount of ice water and dried under vacuum. Analytical data indicate that the water molecules are lost on vacuum drying (Found: C, 38.28; H, 3.84; N, 16.29. [Cu2(C12H12N6)(C5H6O2)][NO3]2 requires C, 38.32; H, 3.80; N, 16.24%). λmax/nm (Nujol) 610; νmax/cm−1 3360 (NH), 1757 (ν1 + ν4 ionic NO3−), 1684, 1657 (CN) and 1020 (py) (Nujol).
[Cu2(pahap − H)(dpa)]2[NO3]4·4H2O (6)..
[Cu2(pahap)(H2O)6][NO3]430 (0.72 g, 1.0 mmol) was dissolved in water (20 cm3), and a solution of 1,3-diamino-2-propanol (Hdpa) (0.18 g, 2.0 mmol) in methanol (5 cm3) was added with warming. Deep green crystals suitable for structural analysis were formed on standing at room temperature. The crystals were filtered off, washed with cold methanol/water and dried in air. (Yield 70%) (Found: C, 29.30; H, 3.87; N, 22.66. [Cu2(C12H12N6)(C3H9N2O)]2[NO3]4·4H2O requires C, 29.22; H, 3.76; N, 22.72%). λmax/nm (Nujol) 670; νmax/cm−1 3460 (H2O), 3298 (NH), 1731 (ν1 + ν4 ionic NO3−), 1686, 1632 (CN) and 1023 (py) (Nujol).
[Cu2(bdpah − H)(NO3)2][NO3] (7)..
A solution of bdpah (0.32 g, 1.0 mmol) in dichloromethane (35 cm3) was added to a solution of excess Cu(NO3)2·3H2O (0.97 g, 4.0 mmol) in methanol (15 cm3) and the mixture was stirred at room temperature for 20 min. The dark green solution was filtered, and the filtrate was allowed to stand at room temperature for several days. Dark green crystals, suitable for X-ray structural determination formed; these were filtered off and air–dried. (Yield 79%) (Found: C, 29.97; H, 2.86; N, 23.63. [Cu2(C16H17N8)(NO3)2][NO3] requires C, 30.29; H, 2.70; N, 24.28%); λmax/nm (Nujol) 568 (sh); νmax/cm−1 3387 (deprotonated NH), 3310 (NH), 1763 and 1737 (ν1 + ν4 bidentate NO3−), 1758 and 1753 (ν1 + ν4 monodentate NO3−), 1730 (ν1 + ν4 ionic NO3−), 1642 (CN) and 1005 (py) (Nujol).
Crystallography
Crystal data and information about the data collection and structural refinement are given in Table 1. Diffraction data for single crystals of 1–3 and 5 were collected using a Bruker SMART CCD diffractometer, equipped with an Oxford Cryostream N2 cooling device,34 with graphite monochromated Mo-Kα radiation. Cell parameters were determined and refined using the SMART software,35a raw frame data were integrated using the SAINT program,35b and the structures were solved using direct methods and refined by full–matrix least squares on F2 using SHELXTL.36,37 Non-hydrogen atoms were refined with anisotropic atomic displacement parameters (adps). For 1 one water molecule in the lattice was modeled as disordered over two sites with occupancies 0.85∶0.15. For 2 the space group cannot be unambiguously determined from systematic absences. The structure can be solved in C2/c, but did not refine well. The solution in Cc gave a dramatic improvement in refinement, and so was carried out in this space group. For 3 the monoclinic space group P1 cannot be unambiguously determined from systematic absences. However a satisfactory refinement of the structure in P1 confirmed this space group assignment. Hydrogen atoms bound to carbon atoms were placed in geometrically calculated positions with isotropic adps 1.2 times that of the parent atom. Hydrogen atoms bound to nitrogen and oxygen atoms were located from difference maps and their coordinates and isotropic adps refined. Hydrogen atoms for 5 were treated in a similar fashion.
| Compound |
1
|
2
|
3
|
5
|
6
|
7
|
| Empirical formula |
C24H32Cu2N16O16.15 |
C26H31Cu2N14O15 |
C26H25Cu2N14O13.5 |
C22H32Cu2N8O13 |
C30H48Cu4N20O18 |
C16H17Cu2N11O9 |
|
M
|
930.14 |
906.75 |
876.68 |
743.64 |
1231.02 |
634.47 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/c |
Cc
|
P
1
|
P21/c |
C2/c |
P
1
|
|
a/Å |
11.3954(7) |
20.938(4) |
9.709(1) |
17.226(3) |
16.965(3) |
10.757(4) |
|
b/Å |
14.062(1) |
14.887(3) |
17.688(2) |
12.260(2) |
16.040(9) |
10.993(3) |
|
c/Å |
22.994(1) |
14.372(3) |
19.155(2) |
14.795(3) |
17.045(3) |
10.590(3) |
|
α/° |
|
|
91.691(4) |
|
|
110.80(2) |
|
β/° |
101.167(4) |
127.43(3) |
95.085(3) |
100.208(3) |
100.72(1) |
99.77(3) |
|
γ/° |
|
|
97.501(3) |
|
|
90.34(3) |
|
U/Å3 |
3614.8(4) |
3557.4(12) |
3245.7(9) |
3075(2) |
4557(2) |
1150.6(6) |
|
T/K |
150(2) |
150(2) |
150(2) |
293(2) |
299(1) |
299(1) |
|
Z
|
4 |
4 |
4 |
4 |
4 |
2 |
|
μ/mm−1 |
1.272 |
1.286 |
1.402 |
1.459 |
1.938 |
1.923 |
| Reflections collected: total, independent, Rint |
24639, 8282, 0.0552 |
21146, 9330, 0.0190 |
39180, 15964, 0.0319 |
31674, 7039, 0.0256 |
5636, 5452, 0.019 |
5569, 5289, 0.028 |
| Final R1, wR2 |
0.0476, 0.0871 |
0.0291, 0.0642 |
0.0375, 0.0794 |
0.0263, 0.0620 |
0.032(R), 0.031(Rw) |
0.044(R), 0.043(Rw) |
The diffraction intensities of single crystals of 6 and 7 were collected with graphite-monochromated Mo-Kα X-radiation using a Rigaku AFC6S diffractometer at 299(1) K and the ω–2θ scan technique. The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods.38,39 All atoms except hydrogens were refined anisotropically. Hydrogen atoms were optimized by positional refinement, with isotropic thermal parameters set 20% greater than those of their bonded partners at the time of their inclusion. However they were fixed for the final round of refinement. Neutral atom scattering factors40 and anomalous-dispersion terms41,42 were taken from the usual sources. All calculations were performed with the teXsan43 crystallographic software package using a PC computer.
CCDC reference number 186/1730.
See http://www.rsc.org/suppdata/dt/a9/a907390f/ for crystallographic files in .cif format.
Results and discussion
Structures
Crystal structure of [Cu2(pahap)2(NO3)(H2O)2][NO3]3·H2O (1)..
The structure of 1 is depicted in Fig. 1, and selected bond distances and angles are listed in Table 2. The two distorted pseudo-octahedral copper(II) ions are bridged by two N–N single bonds (N(3)–N(4) 1.419(3), N(10)–N(9) 1.425(3) Å) in a twisted arrangement with the diazine nitrogens coordinated in the metal equatorial planes and one bidentate nitrate bridging in axial positions. Each ligand provides one pyridine ring coordinating in the equatorial plane of one copper center (N(12) for Cu(1); N(1) for Cu(2)) and another pyridine ring coordinating in the axial position of another copper center (N(6) for Cu(1); N(7) for Cu(2); Cu(1)–N(6) 2.225(3), Cu(2)–N(7) 2.235(3) Å). A water molecule is coordinated to each copper(II) center completing the equatorial coordination (O(1) for Cu(1); O(2) for Cu(2)). The two copper equatorial planes are twisted about the two diazine bond vectors (N(4)–N(3) and N(10)–N(9)), with torsional angles of 55.8° (Cu(1)–N(10)–N(9)–Cu(2)) and 56.4° (Cu(1)–N(4)–N(3)–Cu(2)) and folded into a pseudo–boat conformation by 117.8° as a result of the influence of the axially bridging nitrate via O(102) and O(103), which acts like a basket handle (Cu(1)–O(102) 2.588(2), Cu(2)–O(103) 2.579(2) Å). The Cu(1)–Cu(2) separation is 3.8794(5) Å.
Table 2 Selected bond lengths (Å) and angles (°) for 1
| Cu(1)–N(4) |
1.993(2) |
Cu(2)–O(2) |
2.053(3) |
| Cu(1)–N(12) |
1.996(3) |
Cu(2)–N(7) |
2.235(3) |
| Cu(1)–N(10) |
1.999(3) |
Cu(1)–Cu(2) |
3.8794(5) |
| Cu(1)–O(1) |
2.033(3) |
N(3)–N(4) |
1.419(3) |
| Cu(1)–N(6) |
2.225(3) |
N(9)–N(10) |
1.425(3) |
| Cu(2)–N(1) |
2.001(3) |
Cu(1)–O(102) |
2.588(2) |
| Cu(2)–N(9) |
2.001(3) |
Cu(2)–O(103) |
2.579(2) |
| Cu(2)–N(3) |
2.008(3) |
|
|
| |
|
|
|
| N(4)–Cu(1)–N(12) |
169.65(11) |
N(1)–Cu(2)–N(9) |
167.69(11) |
| N(4)–Cu(1)–N(10) |
89.84(10) |
N(1)–Cu(2)–N(3) |
80.01(11) |
| N(12)–Cu(1)–N(10) |
79.86(10) |
N(9)–Cu(2)–N(3) |
89.36(10) |
| N(4)–Cu(1)–O(1) |
96.84(10) |
N(1)–Cu(2)–O(2) |
92.34(11) |
| N(12)–Cu(1)–O(1) |
93.41(10) |
N(9)–Cu(2)–O(2) |
97.26(11) |
| N(10)–Cu(1)–O(1) |
164.69(11) |
N(3)–Cu(2)–O(2) |
168.92(10) |
| N(4)–Cu(1)–N(6) |
77.65(10) |
N(1)–Cu(2)–N(7) |
112.37(10) |
| N(12)–Cu(1)–N(6) |
103.92(10) |
N(9)–Cu(2)–N(7) |
76.49(10) |
| N(10)–Cu(1)–N(6) |
105.25(10) |
N(3)–Cu(2)–N(7) |
106.50(10) |
| O(1)–Cu(1)–N(6) |
89.67(11) |
O(2)–Cu(2)–N(7) |
83.77(10) |
![Structural representation of the cation in [Cu2(pahap)2(NO3)(H2O)2[NO3]3·2.15H2O (1) (40% probability thermal ellipsoids).](/image/article/2000/DT/a907390f/a907390f-f1.gif) |
| | Fig. 1 Structural representation of the cation in [Cu2(pahap)2(NO3)(H2O)2[NO3]3·2.15H2O (1) (40% probability thermal ellipsoids).
| |
Crystal structure of [Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2)..
The structure of 2 is depicted in Fig. 2, and selected bond distances and angles are listed in Table 3. The dicopper(II) cation consists of two distorted, nominally square-pyramidal copper(II) centers each with two pyridine nitrogens, one diazine nitrogen, and one nitrate oxygen in the equatorial plane, and another diazine nitrogen in the axial position. Other nitrate oxygen atoms O(2) and O(5) are poised above the copper centers to provide weak axial contacts (Cu(1)–O(2) 2.591(3), Cu(2)–O(5) 2.562(3) Å), and so the copper centers are probably best described as distorted octahedral. This arrangement differs substantially from that in 1, where pyridine donor groups occupied one axial position per copper. The two ligands bridge the two copper(II) centers orthogonally in a twisted arrangement, with long, axial contacts to the diazine nitrogens N(2) and N(7) (Cu(1)–N(7) 2.198(3), Cu(2)–N(2) 2.159(3) Å). The angle between the CuN2C2 chelate ring least squares planes is 79.8° around N(7)–N(8) and 82.4° around N(2)–N(3).
Table 3 Selected bond lengths (Å) and angles (°) for 2
| Cu(1)–N(3) |
1.966(3) |
Cu(2)–N(1) |
1.996(3) |
| Cu(1)–N(6) |
1.988(3) |
Cu(2)–N(10) |
2.029(3) |
| Cu(1)–N(5) |
2.021(3) |
Cu(2)–O(4) |
2.081(3) |
| Cu(1)–O(1) |
2.043(2) |
Cu(2)–N(2) |
2.159(3) |
| Cu(1)–N(7) |
2.198(3) |
Cu(2)–O(5) |
2.562(3) |
| Cu(1)–O(2) |
2.591(3) |
N(2)–N(3) |
1.401(5) |
| Cu(1)–Cu(2) |
3.9303(8) |
|
|
| |
|
|
|
| N(3)–Cu(1)–N(6) |
168.71(16) |
N(8)–Cu(2)–N(1) |
171.07(16) |
| N(3)–Cu(1)–N(5) |
80.73(12) |
N(8)–Cu(2)–N(10) |
80.33(13) |
| N(6)–Cu(1)–N(5) |
104.83(12) |
N(1)–Cu(2)–N(10) |
103.48(12) |
| N(3)–Cu(1)–O(1) |
89.15(11) |
N(8)–Cu(2)–O(4) |
91.50(11) |
| N(6)–Cu(1)–O(1) |
89.85(11) |
N(1)–Cu(2)–O(4) |
88.68(11) |
| N(5)–Cu(1)–O(1) |
152.67(12) |
N(10)–Cu(2)–O(4) |
151.86(12) |
| N(3)–Cu(1)–N(7) |
92.06(13) |
N(8)–Cu(2)–N(2) |
93.49(14) |
| N(6)–Cu(1)–N(7) |
77.23(14) |
N(1)–Cu(2)–N(2) |
77.69(14) |
| N(5)–Cu(1)–N(7) |
102.48(12) |
N(10)–Cu(2)–N(2) |
107.37(12) |
| O(1)–Cu(1)–N(7) |
103.19(12) |
O(4)–Cu(2)–N(2) |
99.93(12) |
![Structural representation of the cation in [Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2) (40% probability thermal ellipsoids).](/image/article/2000/DT/a907390f/a907390f-f2.gif) |
| | Fig. 2 Structural representation of the cation in [Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2) (40% probability thermal ellipsoids).
| |
Crystal structure of [Cu2(pahap)(dppn)(H2O)(NO3)][NO3]3 (3)..
Two crystallographically independent, but very similar, molecules have been found in 3. Fig. 3 illustrates the full structure of one of them, and Fig. 4 depicts the expanded view of the coordination cores in both molecules. Selected bond distances and angles are given in Table 4. In each molecule, the ligand dppn adopts a planar structure, as expected, with the same structural features as found in its other complexes,44–47 and the open–chain diazine ligand pahap has a twisted conformation as usual.
Table 4 Selected bond distances (Å) and angles (°) for 3
| Cu(1)–N(7) |
1.935(2) |
Cu(2)–N(3) |
2.228(2) |
Cu(4)–O(2) |
2.055(2) |
Cu(4)–N(15) |
2.022(2) |
| Cu(1)–N(1) |
1.990(2) |
Cu(3)–N(17) |
1.923(2) |
Cu(4)–N(13) |
2.194(2) |
Cu(4)–O(2) |
2.055(2) |
| Cu(1)–N(8) |
2.027(2) |
Cu(3)–N(11) |
1.977(2) |
N(2)–N(3) |
1.334(3) |
Cu(4)–N(13) |
2.194(2) |
| Cu(1)–N(2) |
2.048(2) |
Cu(3)–N(18) |
2.024(2) |
N(6)–N(7) |
1.405(3) |
N(12)–N(13) |
1.334(3) |
| Cu(2)–N(6) |
1.940(2) |
Cu(3)–N(12) |
2.064(2) |
Cu(3)–N(18) |
2.024(2) |
N(16)–N(17) |
1.410(3) |
| Cu(2)–N(4) |
1.988(2) |
Cu(4)–N(16) |
1.935(2) |
Cu(3)–N(12) |
2.064(2) |
Cu(1)–Cu(2) |
3.932(2) |
| Cu(2)–N(5) |
2.018(2) |
Cu(4)–N(14) |
1.980(2) |
Cu(4)–N(16) |
1.935(2) |
Cu(3)–Cu(4) |
3.936(2) |
| Cu(2)–O(1) |
2.032(2) |
Cu(4)–N(15) |
2.022(2) |
Cu(4)–N(14) |
1.980(2) |
|
|
| |
|
|
|
|
|
|
|
| N(7)–Cu(1)–N(1) |
168.94(9) |
N(4)–Cu(2)–N(5) |
99.53(9) |
N(17)–Cu(3)–N(11) |
165.33(9) |
N(14)–Cu(4)–N(15) |
98.14(9) |
| N(7)–Cu(1)–N(8) |
81.04(9) |
N(6)–Cu(2)–O(1) |
96.64(10) |
N(17)–Cu(3)–N(18) |
81.04(9) |
N(16)–Cu(4)–O(2) |
92.00(9) |
| N(1)–Cu(1)–N(8) |
105.01(9) |
N(4)–Cu(2)–O(1) |
87.46(10) |
N(11)–Cu(3)–N(18) |
105.26(9) |
N(14)–Cu(4)–O(2) |
92.92(9) |
| N(7)–Cu(1)–N(2) |
100.04(9) |
N(5)–Cu(2)–O(1) |
154.03(9) |
N(17)–Cu(3)–N(12) |
100.03(9) |
N(15)–Cu(4)–O(2) |
150.19(9) |
| N(1)–Cu(1)–N(2) |
80.68(9) |
N(6)–Cu(2)–N(3) |
92.31(9) |
N(11)–Cu(3)–N(12) |
80.66(9) |
N(16)–Cu(4)–N(13) |
93.81(8) |
| N(8)–Cu(1)–N(2) |
143.66(9) |
N(4)–Cu(2)–N(3) |
78.67(9) |
N(18)–Cu(3)–N(12) |
152.22(9) |
N(14)–Cu(4)–N(13) |
79.13(8) |
| N(6)–Cu(2)–N(4) |
170.28(9) |
N(5)–Cu(2)–N(3) |
113.39(8) |
N(16)–Cu(4)–N(14) |
171.56(9) |
N(15)–Cu(4)–N(13) |
116.67(8) |
| N(6)–Cu(2)–N(5) |
80.60(9) |
O(1)–Cu(2)–N(3) |
92.46(9) |
N(16)–Cu(4)–N(15) |
80.79(9) |
O(2)–Cu(4)–N(13) |
92.53(9) |
![Structural representation of one molecular cation of [Cu2(pahap)(dppn)(NO3)(H2O)][NO3]3 (3) (40% probability thermal ellipsoids).](/image/article/2000/DT/a907390f/a907390f-f3.gif) |
| | Fig. 3 Structural representation of one molecular cation of [Cu2(pahap)(dppn)(NO3)(H2O)][NO3]3 (3) (40% probability thermal ellipsoids).
| |
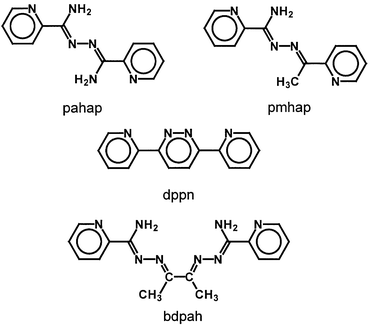
 |
| | Fig. 4 Expanded view showing copper coordination cores and the conformation difference between the two molecules in 3.
| |
The coordination geometries for all copper(II) centers are in between a square pyramid and a trigonal bipyramid, and using the distortion index (τ),48 the values in the range 0.219–0.421 suggest that a distorted square pyramid is the most appropriate stereochemical description in all cases, with short equatorial contacts to an N4 in-plane donor set for Cu(1) and Cu(3), and N3O for Cu(2) and Cu(4). The weak axial coordination positions at Cu(1) and Cu(3) are occupied by nitrate anions (Cu(1)–O(24) 2.536(2); Cu(3)–O(15) 2.629(2) Å), whereas diazine nitrogens from dppn are bonded axially to Cu(2) and Cu(4) (Cu(2)–N(3) 2.228(2), Cu(4)–N(13) 2.194(2) Å). Therefore, within each molecule the two copper(II) centers are bridged by an open-chain diazine unit in the equatorial plane, and by another aromatic diazine unit in an orthogonal manner, to form a boat conformation (boat 1: Cu(1)–N(2)–N(3)–Cu(2)–N(6)–N(7); boat 2: Cu(3)–N(12)–N(13)–Cu(4)–N(16)–N(17)).
The boat conformations in the two molecules differ slightly. The two oxygens (water O(1) and nitrate O(24) ) are located in a trans-position in boat 1, while another two oxygens (water O(15) and nitrate O(2)) are located in a cis-position in boat 2. This results in the dihedral angle between the least squares planes Cu(1)–N(7)–C(21)–C(22)–N(8) and Cu(2)–N(5)–C(19)–C(20)–N(6) (85.9°) being quite different from that between the least squares planes Cu(3)–N(17)–C(47)–C(48)–N(18) and Cu(4)–N(15)–C(45)–C(46)–N(16) (78.8°). The torsion angle around N(6)–N(7) (C(20)–N(6)–N(7)–C(21)) is 104.7°, while that around the N(16)–N(17) bond (C(46)–N(16)–N(17)–C(47)) is only 90.6°.
Crystal structure of [Cu2(pahap)(C2O4)2]·0.5H2O (4)..
A preliminary structural representation for 4 is shown in Fig. 5.29 A weak data set has prevented a full refinement of this structure, but the main features are fully revealed. Two bidentate chelating oxalate groups are bound to the copper centres, forming almost square N2O2 basal planes with Cu-donor distances of <1.98 Å. The Cu–Cu separation is 4.25 Å and the N–N distance (1.41 Å) confirms a single N–N bond bridge. Oxygen atoms from water molecules form weak axial contacts to the coppers (Cu(1)–O(9) 2.51, Cu(2)–O(10) 2.30 Å). There are no apparent hydrogen bonding contacts which could contribute to the degree of twisting of the copper magnetic planes around the N–N bond, and the dihedral angle of 77.9° between the CuN2C2 chelate rings appears to result from steric and crystal packing effects.
![Preliminary structural representation of [Cu2(pahap)(C2O4)2]· 2H2O (4).](/image/article/2000/DT/a907390f/a907390f-f5.gif) |
| | Fig. 5 Preliminary structural representation of [Cu2(pahap)(C2O4)2]· 2H2O (4).
| |
Crystal structure of [Cu2(pahap)(acac)2(H2O)2][NO3]2·H2O (5)..
The structure of 5 is shown in Fig. 6, and selected bond distances and angles are listed in Table 5. The two square-pyramidal copper(II) centers are twisted around the single N–N diazine bond (N(3)–N(4) 1.407(2) Å), each with an N2O2 basal donor set from the bidentate acetylacetonate and a pyridine and a diazine nitrogen. Water molecules complete the axial coordination (Cu(1)–O(3) 2.369(1), Cu(2)–O(6) 2.440(2) Å). The dihedral angle between the CuN2C2 chelate ring least-squares planes is 97.96(4)°, and the twisting of the copper planes is brought about by a combination of steric effects associated mainly with the acetylacetonate groups, and also by a pair of significant hydrogen bonding contacts. The axial water molecules (O(3) and O(6)) are in close proximity to two of the acetylacetonate oxygens (O(4) and O(2) respectively; O(2)–O(6) 2.809(2); O(3)–O(4) 2.926(2) Å; O(6)–H(62w)–O(2) 173.9(3); O(3)–H(32w)–O(4) 162.6(3)°), and the resulting hydrogen bonds effectively lock the conformation of the molecule. This leads to quite a large copper–copper separation (4.360(1) Å).
Table 5 Selected bond distances (Å) and angles (°) relevant to the copper coordination spheres in 5
| Cu(1)–O(1) |
1.9142(12) |
Cu(2)–O(4) |
1.9461(13) |
| Cu(1)–O(2) |
1.9314(13) |
Cu(2)–N(4) |
1.9974(14) |
| Cu(1)–N(3) |
1.9766(14) |
Cu(2)–N(6) |
2.018(2) |
| Cu(1)–N(1) |
2.004(2) |
N(3)–N(4) |
1.407(2) |
| Cu(1)–O(3) |
2.3686(14) |
Cu(1)–Cu(2) |
4.360(1) |
| Cu(2)–O(5) |
1.9189(13) |
|
|
| |
|
|
|
| O(1)–Cu(1)–O(2) |
93.83(5) |
N(3)–Cu(1)–N(1) |
79.71(6) |
| O(1)–Cu(1)–N(3) |
166.31(6) |
O(1)–Cu(1)–O(3) |
99.92(5) |
| O(2)–Cu(1)–N(3) |
92.73(6) |
O(2)–Cu(1)–O(3) |
92.94(5) |
| O(1)–Cu(1)–N(1) |
92.43(6) |
N(3)–Cu(1)–O(3) |
91.72(6) |
| O(2)–Cu(1)–N(1) |
170.58(6) |
N(1)–Cu(1)–O(3) |
92.89(5) |
| |
![Structural representation of the cation in [Cu2(pahap)(acac)2(H2O)2][NO3]2·H2O (5) (40% probability thermal ellipsoids).](/image/article/2000/DT/a907390f/a907390f-f6.gif) |
| | Fig. 6 Structural representation of the cation in [Cu2(pahap)(acac)2(H2O)2][NO3]2·H2O (5) (40% probability thermal ellipsoids).
| |
Crystal structure of [Cu2(pahap − H)(dpa)]2[NO3]4·4H2O (6)..
The structure of the cation in 6 is shown in Fig. 7, and selected bond distances and angles are listed in Table 6. The tetranuclear, rectangular structural framework can be considered as two dinuclear parts, in which two copper(II) centres are bridged in an almost trans fashion (Cu(1)–N(3)–N(4)–Cu(2) torsion angle 175.7°) by a deprotonated pentadentate pahap ligand, and then the two parts are linked by a bridging, deprotonated dpa fragment. The Cu(1) centres have square-pyramidal geometries, with short in plane contacts (<2.03 Å). The Cu(1)–N(5) distance (1.937(3) Å) is very short and indicative of proton loss at N(5). The long contact to O(1) (2.252(2) Å) defines the axial ligand of the square-pyramid. Cu(2) has a distorted square geometry, with no significant axial contacts. All copper donor atom distances are short (<1.99 Å), with a very short contact to O(1) (1.894(2) Å) consistent with a deprotonated alkoxide fragment (Cu(1)–O(1)–Cu(2) 111.15(8)°).
Table 6 Selected bond distances (Å) and angles (°) for 6
| Cu(1)–O(1) |
2.252(2) |
Cu(2)–N(4) |
1.984(2) |
| Cu(1)–N(1) |
2.025(3) |
Cu(2)–N(7) |
1.991(3) |
| Cu(1)–N(3) |
1.950(2) |
Cu(2)–N(8) |
1.966(2) |
| Cu(1)–N(5) |
1.937(3) |
Cu(1)–Cu(2) |
4.847(2) |
| Cu(1)–N(6) |
2.021(2) |
Cu(1)–Cu(2)′ |
3.425(2) |
| Cu(2)–O(1) |
1.894(2) |
N(3)–N(4) |
1.400(3) |
| |
|
|
|
| O(1)–Cu(1)–N(1) |
90.27(8) |
N(5)–Cu(1)–N(6) |
97.7(1) |
| O(1)–Cu(1)–N(3) |
103.17(8) |
O(1)–Cu(2)–N(4) |
102.86(8) |
| O(1)–Cu(1)–N(5) |
102.72(9) |
O(1)–Cu(2)–N(7) |
86.6(1) |
| O(1)–Cu(1)–N(6) |
81.71(8) |
O(1)–Cu(2)–N(8) |
150.60(9) |
| N(1)–Cu(1)–N(3) |
80.33(9) |
N(4)–Cu(2)–N(7) |
152.0(1) |
| N(1)–Cu(1)–N(5) |
157.7(1) |
N(4)–Cu(2)–N(8) |
83.09(9) |
| N(1)–Cu(1)–N(6) |
102.1(1) |
N(7)–Cu(2)–N(8) |
101.6(1) |
| N(3)–Cu(1)–N(5) |
79.12(9) |
Cu(1)–O(1)–Cu(2) |
111.15(8) |
| N(3)–Cu(1)–N(6) |
174.6(1) |
|
|
![Structural representation of the tetranuclear cation in [Cu2(pahap − H)(dpa)]2[NO3]4·4H2O (6) (40% probability thermal ellipsoids).](/image/article/2000/DT/a907390f/a907390f-f7.gif) |
| | Fig. 7 Structural representation of the tetranuclear cation in [Cu2(pahap − H)(dpa)]2[NO3]4·4H2O (6) (40% probability thermal ellipsoids).
| |
Crystal structure of [Cu2(bdpah − H)(NO3)2][NO3] (7)..
The structure of 7 is shown in Fig. 8 and consists of a deprotonated hexadentate ligand bridging two copper(II) centres by a single N–N bond (N(5)–N(6) 1.392(5) Å). Selected bond distances and angles are given in Table 7. Cu(1) is bonded to four nitrogen atoms in a square planar geometry, including N(7) which has a single proton, and is an anionic ligand site (Cu(1)–N(7) (1.934(4) Å). Cu(2) is bound to two ligand nitrogen sites and two monodentate nitrate oxygens (O(1) and O(4)) with short (<2.03 Å) contacts, but a more distant axial contact to O(2) (2.496(6) Å) suggests that one nitrate is bidentate and that the geometry at Cu(2) is distorted square-pyramidal. The copper–copper separation of 4.511(2) Å is consistent with the moderately large Cu–N–N–Cu torsional angle of 130.3°, which implies a significant bending of the copper square planes about the N–N bridge. C(11) and N(5) have essentially trigonal planar character, while N(6) has a significant pyramidal distortion (solid angle at N(6) 347.2°), indicating that the bend occurs at this atom.
Table 7 Selected bond distances (Å) and angles (°) for 7
| Cu(1)–N(1) |
1.990(4) |
Cu(2)–N(6) |
1.974(4) |
| Cu(1)–N(3) |
1.905(4) |
Cu(2)–N(8) |
1.977(4) |
| Cu(1)–N(5) |
1.958(4) |
N(5)–N(6) |
1.392(5) |
| Cu(1)–N(7) |
1.934(4) |
N(6)–C(11) |
1.345(6) |
| Cu(2)–O(1) |
2.021(4) |
N(7)–C(11) |
1.292(5) |
| Cu(2)–O(4) |
1.971(4) |
N(3)–N(4) |
1.374(5) |
| |
|
|
|
| N(1)–Cu(1)–N(3) |
82.3(2) |
O(1)–Cu(2)–O(4) |
92.5(2) |
| N(1)–Cu(1)–N(5) |
170.8(2) |
O(1)–Cu(2)–N(6) |
93.6(2) |
| N(1)–Cu(1)–N(7) |
106.7(2) |
O(1)–Cu(2)–N(8) |
157.8(2) |
| N(3)–Cu(1)–N(5) |
90.3(2) |
O(4)–Cu(2)–N(6) |
171.5(2) |
| N(3)–Cu(1)–N(7) |
168.7(2) |
O(4)–Cu(2)–N(8) |
95.3(2) |
| N(5)–Cu(1)–N(7) |
81.4(2) |
N(6)–Cu(2)–N(8) |
81.2(2) |
![Structural representation of the cation in [Cu2(bdpah − H)(NO3)2][NO3] (7) (40% probability thermal ellipsoids).](/image/article/2000/DT/a907390f/a907390f-f8.gif) |
| | Fig. 8 Structural representation of the cation in [Cu2(bdpah − H)(NO3)2][NO3] (7) (40% probability thermal ellipsoids).
| |
Spectroscopy
Infrared bands associated with the NH2 groups and lattice and coordinated water are observed for these complexes in the range 3560–3200 cm−1. The absence of an absorption associated with νOH for 6 supports proton loss from O(1) (Fig. 7). In general strong νCN bands are observed above 1630 cm−1, higher in energy than those of the free ligands (νCN pahap, pmhap, bdpah; 1608, 1613, 1613 cm−1 respectively), and in agreement with the fact that in most cases these ligands adopt a twisted conformation in the complexes.30 The free ligands themselves have flat structures with significant intramolecular conjugation, which is broken when the ligands are twisted. Characteristic (ν1 + ν4) nitrate bands49 are observed in the region 1700–1800 cm−1 in general agreement with the role of the nitrate groups observed in the structures. Pyridine ring breathing bands are found at 1005 cm−1 or higher in all the complexes,50 in agreement with the fact that all pyridine rings are coordinated.
Solid state Nujol mull transmittance electronic spectra for 1–6 are quite similar, with one broad visible band observed in each case in the range 610–720 nm, consistent with the square or effectively five-coordinate geometries observed at the copper(II) centers. Aqueous solution spectra are slightly different from their solid state spectra, suggesting minor changes to the coordination environment in solution. Complex 7 has a higher energy absorption in the solid state consistent with the square-planar copper centre (Cu(1)).
Magnetism
Variable temperature magnetic susceptibility measurements were carried out on powdered samples of all of the complexes, taken from the same uniform batches used for structural determinations, in the temperature range 3–300 K. The room temperature magnetic moment for complex 1 (1.94 μB) is close to the normal value for an uncoupled copper(II) system, and might suggest the absence of spin exchange. The variable temperature susceptibility data were fitted to the Bleaney–Bowers equation51 (eqn. (1); J is the exchange integral, ρ is the fraction of paramagnetic impurity, θ is a Weiss-like corrective term, Nα is the temperature independent paramagnetism, and other terms have their usual meaning; the equation is based on the Hamiltonian H = −2JS1·S2) to give g = 2.191(7), 2J = −0.4(3) cm−1, ρ = 0.00046, Nα = 75 × 10−6 cm3 mol−1 (Cu), θ = −2.1 K, 102R = 2.2 (R = [Σ(χobs − χcalc)2/Σχobs2]1/2). Since the singlet–triplet splitting is very small, and possibly comparable with the Zeeman energy (gβH), the data were also fitted to the magnetization expression (eqn. (2), (3) and (4)), corrected for interdimer exchange (zJ′).52,53
| |

|
(1)
|
| | |
M = Ngβ(sinh(gβH/kT))/(exp(−2J/kT) + 2cosh(gβH/kT + 1)
|
(2)
|
| | |
χ′Cu = χCu/(1 − (2zJ′χCu/Ng2β2))
|
(4)
|
A comparable fit was obtained by this approach with g = 2.19(1), 2J = −2.2(5) cm−1, zJ′ = −1.6 cm−1, Nα = 60 × 10−6 cm3 mol−1 (Cu), ρ = 0.00046, thus confirming the very weak antiferromagnetic exchange in addition to a weak, intermolecular, antiferromagnetic component. The structure did not however reveal any significant intermolecular contacts. The very weak intramolecular spin coupling is associated with the small torsional angles (Cu(2)–N(9)–N(10)–Cu(1) 54.95°; Cu(2)–N(3)–N(4)–Cu(1) 54.47°) defining the rotation of the copper planes about the two diazine bonds in agreement with the results of our previous studies.30,31
The compound [Cu2(pmhap)2(NO3)2][NO3]2·3H2O (2) also has a high room temperature magnetic moment (1.91 μB), which is essentially constant throughout the 5–300 K temperature range, indicative of no net coupling between the copper(II) centers. This clearly results because the two copper(II) centers are bridged orthogonally by the two N–N single bonds (Fig. 2).
The compound [Cu2(pahap)(dppn)(H2O)(NO3)[NO3]3 (3) has a slightly lower room temperature magnetic moment (1.74 μB), and a plot of χmversus temperature reveals a maximum in the susceptibility at about 40 K, indicative of dominant antiferromagnetic exchange. The data were fitted to eqn. (1) with g = 2.04(2), 2J = −32.5(7) cm−1, ρ = 0.0095, Nα = 61 × 10−6 cm3 mol−1 (Cu), θ = −24 K, 102R = 1.9 (R = [Σ(χobs − χcalc)2/Σχobs2]1/2). The necessity for inclusion of a large Weiss–like corrective temperature term (θ) indicates that the model is inappropriate, but suggests that additional exchange components are likely to be present, and that they cannot be considered to result from minor intermolecular exchange effects. The absence of significant intermolecular connections that could contribute to exchange coupling, and the presence of two quite different molecules in the unit cell, with very different twist angles of the copper magnetic planes about the N–N bonds suggests that a dinuclear model with two J values might be more appropriate. The fitting of the data to such a model was not well behaved, and somewhat unstable, but gave two quite different 2J values (≈−85 cm−1 and ≈ −30 cm−1), with poorer R values than before. This result is consistent with the structure in which the combination of two different dinuclear species with quite different dihedral angles between the copper magnetic planes would be expected to have quite different J values, based on our previous studies.30,31
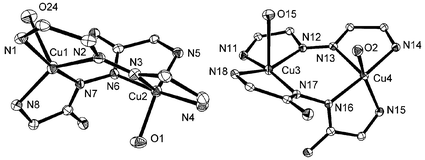
The magnetic moment for 4 is greater than 1.8 μB above 50 K, and drops to 1.3 μB at 3 K (Fig. 9), indicating the possible presence of very weak antiferromagnetic coupling. The variable temperature magnetic moment data were fitted successfully to eqn. (1), and the best fit of the data is represented by the solid line in Fig. 9, which was calculated for g = 2.124(8) cm−1, 2J = −4.4(2) cm−1, θ = −0.5 K, ρ = 0.00001, and Nα = 60 × 10−6 cm3 mol−1 (Cu), 102R = 0.61. This result is entirely consistent with our previous studies30,31 showing that at dihedral angles close to 80° antiferromagnetic spin exchange should be very weak or close to zero.
 |
| | Fig. 9 Variable temperature magnetic data for complex 4. The solid line was calculated from eqn. (1) with g = 2.124(8), 2J = −4.4(2) cm−1, ρ = 0, Nα = 60 × 10−6 cm3 mol−1, θ = −0.5 K (102R = 0.61). | |
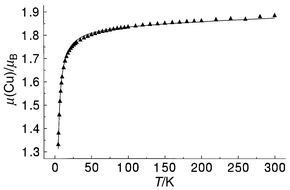
Variable temperature magnetic susceptibility data for 5 (Fig. 10) reveal a maximum in susceptibility at ≈70 K, indicating significant intramolecular antiferromagnetic coupling. A good fit to eqn. (1) was obtained with g = 2.17(1), 2J = −69.7(5) cm−1, ρ = 0.00212, θ = 0 K, Nα = 60 × 10−6 cm3 mol−1 (Cu), 102R = 1.8. The solid line in Fig. 10 was calculated with these parameters. The comparatively large −2J value for this compound is entirely consistent with the fact that as the ligand opens up by rotation around the N–N single bond, enhanced nitrogen p orbital overlap occurs leading to enhanced antiferromagnetic coupling between the copper centres.31 In this case the rotation is effectively locked by the two hydrogen bonding contacts (Fig. 6) from oxygens O(3) and O(6) to O(4) and O(2) respectively (vide ante).
 |
| | Fig. 10 Variable temperature magnetic data for complexes 5–7. The solid line for 5 was calculated from eqn. (1), with g = 2.17(1), 2J = −69.7(5) cm−1, ρ = 0.00212, Nα = 60 × 10−6 cm3 mol−1 and θ = 0 K (102R = 1.8). The solid line for 6 was calculated from eqn. (1) with g = 2.24(1), 2J = −186.4(5) cm−1, ρ = 0.00002, Nα = 25 × 10−6 cm3 mol−1, θ = 0 K (102R = 0.10). The solid line for 7 was calculated from eqn. (1) with g = 2.050(2), 2J = −112.0(1) cm−1, ρ = 0.022, Nα = 90 × 10−6 cm3 mol−1, θ = 0 K (102R = 0.60). | |
Variable temperature magnetic data for 6 show a maximum in susceptibility (Fig. 10) at approximately 160 K, indicative of quite strong antiferromagnetic exchange. Since the connection between the two halves of the molecule via the alkoxide bridge is orthogonal there can theoretically be no antiferromagnetic coupling between Cu(1) and Cu(2) through this bridge. In such a case the magnetic data can therefore be fitted to eqn. (1). An excellent fit of the data was obtained to eqn. (1), with g = 2.24(1), 2J = −186.4(5) cm−1, θ = 0 K, ρ = 0.00002, Nα = 25 × 10−6 cm3 mol−1 (Cu) (102R = 0.10). The solid line in Fig. 10 was calculated with these parameters. The spin coupling in 6 is clearly dominated by the N–N single bond bridge, and the large −2J value is entirely consistent with the previous correlations between rotation angle of the copper magnetic planes around the flexible diazine bridge.31
A plot of magnetic susceptibility against temperature for 7 is also illustrated in Fig. 10, and displays a maximum at ≈100 K, indicating moderately strong antiferromagnetic coupling between the copper(II) centres. The data gave an excellent fit to eqn. (1) with g = 2.050(2), 2J = −112.0(1) cm−1, θ = 0 K, ρ = 0.022, Nα = 90 × 10−6 emu (Cu) (102R = 0.6). The solid line in Fig. 10 was calculated with these parameters. The exchange integral is much smaller than that obtained for 6, which is consistent with the smaller Cu–N–N–Cu torsional angle and the previous magnetostructural correlation.30,31
Conclusion
A series of new 2∶1 and 2∶2 dicopper(II) complexes with N–N single bond bridges has been studied, in addition to a novel tetranuclear cluster. Two complexes with double N–N bridges have acutely twisted structures and are essentially ‘non-coupled’, either as a result of strict orthogonality between the copper magnetic orbitals, or accidental orthogonality resulting from the critical twist angle around the N–N bond. The other systems involve constrained rotations of the copper magnetic planes about a single N–N bond at larger angles, due to additional bridging and hydrogen bonding or steric interactions, and the exchange coupling becomes antiferromagnetic, consistent with previous magnetostructural correlations. This study provides additional examples of dicopper(II) complexes bridged by N–N single bonds, where magnetic properties can be effectively tuned by the simple concept of rotation of the copper magnetic planes about this bond.
Acknowledgements
We thank the Natural Sciences and Engineering Council of Canada (NSERC), EPSRC (UK), NATO and Monbusho (Japan) for financial support for this study.
References
- S. S. Tandon, L. K. Thompson and R. C. Hynes, Inorg. Chem., 1992, 31, 2210 CrossRef CAS.
- R. Prins, P. J. M. W. L. Birker, J. G. Haasnoot, G. C. Verschoor and J. Reedijk, Inorg. Chem., 1985, 24, 4128 CrossRef CAS.
- L. Chen, L. K. Thompson and J. N. Bridson, Inorg. Chem., 1993, 32, 2938 CrossRef CAS.
- S. S. Tandon, L. K. Thompson, M. E. Manuel and J. N. Bridson, Inorg. Chem., 1995, 34, 2356 CrossRef.
- T. Kamiusuki, H. Ōkawa, N. Matsumoto and S. Kida, J. Chem. Soc., Dalton Trans., 1990, 195 RSC.
- J. C. Bayon, P. G. Esteban, P. G. Rasmussen, K. N. Baker, C. W. Hahn and M. M. Gumz, Inorg. Chem., 1991, 30, 2572 CrossRef CAS.
- J. Pons, X. López, J. Casabó, F. Teixidor, A. Caubet, J. Ruis and C. Miravitlles, Inorg. Chim. Acta, 1992, 195, 61 CrossRef CAS.
- A. Bencini, D. Gatteschi, C. Zanchini, J. G. Haasnoot, R. Prins and J. Reedijk, Inorg. Chem., 1992, 24, 2812.
- W. M. E. Koomen-van Oudenniel, R. A. G. de Graff, J. Haasnoot, R. Prins and J. Reedijk, Inorg. Chem., 1989, 28, 1128 CrossRef CAS.
- P. J. van Koningsbruggen, J. G. Haasnoot, R. A. G. de Graff, J. Reedijk and S. Slingerland, Acta Crystallogr., Sect. C, 1992, 48, 1923 CrossRef.
-
Z. Xu
, Ph.D. Thesis, Memorial University of Newfoundland, 1998.
Search PubMed.
-
(a) W. J. Stratton and D. H. Busch, J. Am. Chem. Soc., 1960, 82, 4834 CrossRef CAS;
(b) W. J. Stratton and D. H. Busch, J. Am. Chem. Soc., 1958, 80, 1286 CrossRef CAS;
(c) W. J. Stratton and D. H. Busch, J. Am. Chem. Soc., 1958, 80, , 3191 CrossRef.
- W. J. Stratton and P. J. Ogren, Inorg. Chem., 1970, 9, 2588 CrossRef CAS.
- P. D. W. Boyd, M. Gerloch and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1974, 1097 RSC.
- J. Saroja, V. Manivannan, P. Chakraborty and S. Pal, Inorg. Chem., 1995, 34, 3099 CrossRef CAS.
- C. J. O’Connor, R. J. Romananch, D. M. Robertson, E. E. Eduok and F. R. Fronczek, Inorg. Chem., 1983, 22, 449 CrossRef CAS.
- T. C. Woon, L. K. Thompson and P. Robichaud, Inorg. Chim. Acta, 1984, 90, 201 CrossRef CAS.
- P. Souza, A. I. Matesanz and V. Fernández, J. Chem. Soc., Dalton Trans., 1996, 3011 RSC.
- J. García-Jojal, J. García-Jaca, R. Cortés, T. Rojo, M. K. Urtiaga and M. I. Arriortua, Inorg. Chim. Acta, 1996, 249, 25 CrossRef.
- A. Mangia, C. Pelizzi and G. Pelizzi, Acta Crystallogr., Sect. B, 1974, 30, , 2146 CrossRef.
- A. Bonardi, S. Ianelli, C. Pelizzi and G. Pelizzi, Inorg. Chim. Acta, 1991, 187, 167 CrossRef CAS.
- A. Bacchi, A. Bonini, M. Carcelli, F. Ferraro, E. Leporati, C. Pelizzi and G. Pelizzi, J. Chem. Soc., Dalton Trans., 1996, 2699 RSC.
- A. E. Koziol, R. C. Palenik and G. J. Palenik, J. Chem. Soc., Chem. Commun., 1989, 650 RSC.
- M. Lagrenée, S. Sueur and J. P. Wignacourt, Acta Crystallogr., Sect. C, 1991, 47, 1158 CrossRef.
- A. Bacchi, L. P. Battaglia, M. Carcelli, C. Pelizzi, G. Pelizzi and C. Solinas and M.A. Zoroddu, J. Chem. Soc., Dalton Trans., 1993, 775 RSC.
- E. W. Ainscough, A. M. Brodie, J. D. Ranford and J. M. Waters, Inorg. Chim. Acta, 1995, 236, 83 CrossRef CAS.
- X. Chen, S. Zhan, C. Hu, Q. Meng and Y. Liu, J. Chem. Soc., Dalton Trans., 1997, 245 RSC.
- P. J. van Koningsbruggen, E. Muller, J. G. Haasnoot and J. Reedijk, Inorg. Chim. Acta, 1993, 208, 37 CrossRef CAS.
-
Z. Xu,
C. J. Matthews, L. K. Thompson and
S. L. Heath,
unpublished work..
- Z. Xu, L. K. Thompson and D. O. Miller, Inorg. Chem., 1997, 36, 3985 CrossRef CAS.
- L. K. Thompson, Z. Xu, A. E. Goeta, J. A. K. Howard, H. J. Clase and D. O. Miller, Inorg. Chem., 1998, 37, 3217 CrossRef CAS.
- D. S. Brown, V. H. Crawford, J. W. Hall and W. E. Hatfield, J. Phys. Chem., 1977, 81, 1303 CrossRef CAS.
- W. H. Butte and F. H. Case, J. Org. Chem., 1961, 26, 4690 CrossRef CAS.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105 CrossRef CAS.
-
(a)
Siemens, SMART Data Collection Software, Ver. 4.050, Siemens Analytical X-ray Instruments Inc., Madison, WI, 1996;
Search PubMed;
(b)
Siemens, SAINT Data Reduction Software, Version 4.050, Siemens Analytical X-ray Instruments Inc., Madison, WI, 1996.
Search PubMed.
-
G. M. Sheldrick
, SHELXTL 5.04/VMS, An integrated system for solving, refining and displaying crystal structures from diffraction data, Siemens Analytical X-ray Instruments Inc., Madison, WI, 1995.
Search PubMed.
-
G. M. Sheldrick
, SADABS, Empirical Absorption Correction Program, University of Göttingen, 1996.
Search PubMed.
- SIR92: A. Altomare, M. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- DIRDIF94:
P. T. Beurskens
, G. Admiraal
, G. Beurskens
, W. P. Bosman
, R. de Gelder
, R. Israel
and J. M. M. Smits
, The DIRDIF-94 program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, The Netherlands, 1994.
Search PubMed.
-
D. T. Cromer
and J. T. Waber
, International Tables for X-ray Crystallography, The Kynoch Press, Birmingham, 1974, Vol. IV, Table 2.2A.
Search PubMed.
- J. A. Ibers and W. C. Hamilton, Acta Crystallogr., 1964, 17, 781 CrossRef.
-
D. C. Creagh
and W. J. McAuley
, International Tables for Crystallography, ed. A. J. C. Wilson, Kluwer Academic Publishers, Boston, vol. C, 1992, Table 4.2.6.8, pp. 219–222.
Search PubMed.
-
teXsan for Windows: Crystal Structure Analysis Package, Molecular Structure Corporation, The Woodlands, TX, 1997.
Search PubMed.
- G. De Munno, G. Denti and P. Dapporto, Inorg. Chim. Acta, 1983, 74, 199 CrossRef CAS.
- G. De Munno and G. Denti, Acta Crystallogr., Sect. C, 1984, 40, 616 CrossRef.
- G. De Munno and G. Bruno, Acta Crystallogr., Sect. C, 1984, 40, 2022 CrossRef.
- P. Dapporto, G. De Munno, A. Sega and C. Meali, Inorg. Chim. Acta, 1984, 83, 171 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- A. B. P. Lever, E. Mantovani and B. S. Ramaswamy, Can. J. Chem., 1971, 49, 1957 CAS.
- A. R. Katritzky and A. R. Hands, J. Chem. Soc., 1958, 2202 RSC.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, A, 1952, 214, 451 Search PubMed.
- B. E. Myers, L. Berger and S. Friedberg, J. Appl. Phys., 1969, 40, 1149 CrossRef CAS.
- W. E. Marsh, K. C. Patel, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1983, 22, 511 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.