Synthesis and characterisation of new platinum–acetylide complexes containing diimine ligands
Christopher J.
Adams
*a,
Stuart L.
James
a,
Xiaoming
Liu
b,
Paul R.
Raithby
*a and
Lesley J.
Yellowlees
*b
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW
bDepartment of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, UK EH9 3JJ
First published on 24th December 1999
Abstract
A new class of platinum–bipyridyl compounds has been synthesized by the dehydrohalogenative reaction of [4,4′-bis(tert-butyl)-2,2′-bipyridyl]platinum dichloride [PtCl2(tBu2bipy)] 1 with terminal alkynes HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR, in the presence of copper(I) iodide and diisopropylamine. The products [Pt(CCR)2(tBu2bipy)] (R = C6H4NO2-p2, C6H53, C6H4CH3-p4 or SiMe35), have been characterised by spectroscopic and analytical methods, and a single crystal molecular structure determination has been carried out on 4. Extended Hückel molecular orbital calculations have also been carried out, and the results are used to help rationalise the voltammetric, EPR and spectroelectrochemical properties of the new compounds. These show that compounds 3, 4 and 5 undergo a one-electron bipyridyl based redox process, but that 2 has an unresolved two-electron process located on the nitro groups.
CR, in the presence of copper(I) iodide and diisopropylamine. The products [Pt(CCR)2(tBu2bipy)] (R = C6H4NO2-p2, C6H53, C6H4CH3-p4 or SiMe35), have been characterised by spectroscopic and analytical methods, and a single crystal molecular structure determination has been carried out on 4. Extended Hückel molecular orbital calculations have also been carried out, and the results are used to help rationalise the voltammetric, EPR and spectroelectrochemical properties of the new compounds. These show that compounds 3, 4 and 5 undergo a one-electron bipyridyl based redox process, but that 2 has an unresolved two-electron process located on the nitro groups.
There has been considerable recent interest in transition metal organometallic compounds that contain two acetylide ligands in a cis configuration, due to their suitability as molecular tweezers for the co-ordination of another transition metal fragment in order to form homo- or hetero-bimetallic molecules. Interest arises not only because of their structure, but also their possible catalytic activity and suitability as models for catalytic systems. The complexes are mainly based either upon the bis(cyclopentadienyl)titanium system, or upon platinum systems with tertiary phosphines or C6F5− as the auxiliary ligands.1,2
There is also interest in platinum–bipyridyl complexes, either with regard to their solid state structure and how this is related to their luminescence properties,3 the formation of d9 metal centres,4 or their ability to undergo oxidative addition to form platinum(IV) complexes.5 An underlying problem has been the relative insolubility of platinum–bipyridyl complexes in organic solvents, and recent work has started to utilise substituted bipyridines to overcome this problem.6
We report here the synthesis and characterisation of a new class of platinum–bipyridyl compounds, containing a substituted bipyridyl ligand and σ-bonded acetylides in the remaining co-ordination sites. A preliminary communication on related work has appeared,7 and since this work was started a report containing another platinum–acetylide complex with a diimine ligand has been published.8 We know of only four other published platinum–acetylide structures which also contain nitrogen donors, three of which are square planar platinum(II) compounds such as those detailed here,2,9 and one of which is an octahedral platinum(IV) compound.10
Results and discussion
Synthesis of the compounds
The diimine ligand employed in this study is 4,4′-bis(tert-butyl)-2,2′-bipyridyl (tBu2bipy), which has the chelating properties of unsubstituted bipyridine but confers much greater solubility in organic solvents upon metal complexes. It was synthesized from 4-tert-butylpyridine, not by the generally used method involving Raney nickel![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 11 but by the lesser known but more convenient method of McGill.12 Synthesis of the platinum starting material [PtCl2(tBu2bipy)] 1 from this was relatively straightforward; although tBu2bipy is insoluble in water, the method of Morgan and Burstall13 involving refluxing the ligand with potassium tetrachloroplatinate in dilute hydrochloric acid still works. However, we found that a more convenient synthesis is to displace the acetonitrile ligands from bis(acetonitrile)platinum dichloride, formed in situ from the reaction of platinum dichloride and acetonitrile.14
11 but by the lesser known but more convenient method of McGill.12 Synthesis of the platinum starting material [PtCl2(tBu2bipy)] 1 from this was relatively straightforward; although tBu2bipy is insoluble in water, the method of Morgan and Burstall13 involving refluxing the ligand with potassium tetrachloroplatinate in dilute hydrochloric acid still works. However, we found that a more convenient synthesis is to displace the acetonitrile ligands from bis(acetonitrile)platinum dichloride, formed in situ from the reaction of platinum dichloride and acetonitrile.14
From 1, the synthesis of the acetylide complexes was as previously described.7 Stirring overnight with an excess of acetylene in the presence of a catalytic amount of copper(I) iodide and a small amount of diisopropylamine leads to formation of the acetylide complexes [Pt(CCR)2(tBu2bipy)] 2–5 in good yield. Purification is easily effected by alumina column chromatography (for 3–5) and/or recrystallisation, leading to isolation of the products as air stable yellow solids (Table 1).
| Compound | R | Colour |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (CC)/cm−1 (CC)/cm−1 |
Yield (%) |
|---|---|---|---|---|
| 2 | C6H4NO2-p | Yellow | 2111, 2123 | 54 |
| 3 | C6H5 | Yellow | 2115, 2124 | 83 |
| 4 | C6H4CH3-p | Yellow | 2114, 2126 | 69 |
| 5 | SiMe3 | Yellow-green | 2040, 2056 | 57 |
Characterisation of the complexes
All the compounds show characteristic 1H NMR spectra which are based upon that of free tBu2bipy. The aromatic protons H5 and H6 both show a downfield shift upon complexation, whereas H3 moves upfield slightly. There is a relatively small variation in the spectra as the nature of the acetylide group changes, with the exception of compound 2. The compounds also all show the two ν(CC) bands in their IR spectrum expected for a cis-bis(acetylide) complex, although they differ from similar phosphine complexes in that the more intense of the two bands is the lower energy and that they are at a slightly higher frequency.15
The UV/visible spectra of compounds 1–5 in the 250–1000 nm region have been recorded (Table 2). All except 2 show similar spectra consisting of two major bands (ε ≈ 104 dm3 mol−1 cm−1), one at 380 to 400, and one at 280 to 290 nm. These show a solvatochromic shift, moving to shorter wavelengths with increasing solvent polarity, and, in common with Gidney et al.,16 the bands have been assigned to the first and second metal to ligand charge transfer (MLCT) respectively. These correspond to excitation of an electron from the highest occupied molecular orbital (HOMO) (mainly platinum dx2 − y2 in nature) to the lowest and second lowest unoccupied molecular orbitals (LUMO and SLUMO), which are mainly bipyridyl in nature. The spectra of 1 and 5 also show a double transition at 307 and 320 nm that is not solvatochromic; this may be assigned to a bipyridyl π to π* transition. It appears as a shoulder on the second MLCT band of 4.
| Solvent | ||||
|---|---|---|---|---|
| Compound | R | Aceto-nitrile | Dichloro-methane | Ethyl acetate |
| 2 | C6H4NO2-p | 277 (1.5) | 283 (1.4) | 286 (1.5) |
| 371 (2.9) | 369 (2.3) | 369 (2.5) | ||
| 3 | C6H5 | 284 (3.0) | 284 (4.6) | 287 (4.0) |
| 384 (0.5) | 395 (1.2) | 407 (0.7) | ||
| 4 | C6H4CH3-p | 282 (5.0) | 286 (7.9) | 287 (4.7) |
| 388 (0.9) | 399 (1.1) | 412 (0.7) | ||
| 5 | SiMe3 | 282 (9.1) | 287 (2.4) | 290 (3.5) |
| 375 (2.6) | 384 (0.7) | 398 (0.9) | ||
The spectrum of compound 2 is slightly different. The 280 nm MLCT band is still visible, as is some structure due to the bipyridyl π to π* transition at around 320 nm. However, there is now one dominant absorption at around 370 nm that is much less solvatochromic than the first MLCT band of compounds 3–5. The band has an absorption coefficient approximately four times that of the other complexes, and this spectral difference indicates that the origin of this band may be different from that in 3–5.
A single crystal of complex 4·3CHCl3 suitable for an X-ray diffraction study was grown by evaporation of a chloroform–hexane solution. This confirmed the proposed geometry of the compound, revealing it to be essentially planar (apart from the two tert-butyl groups) (Fig. 1). The average length of the Pt–C bond is 1.944 Å, a little shorter than is generally found in bis(acetylide) complexes of platinum with phosphine ligands17 but in good agreement with the other published bis(acetylide) structure containing a diimine ligand.8 Conversely, the average carbon–carbon distance in the triple bond of 1.23(2) Å is longer than is generally seen. The two acetylide groups bend away from each other, with the deviation from linearity at the acetylenic carbons averaging around 13°. Selected bond lengths and angles are presented in Table 3. There are hydrogen-bonding interactions between the chloroform solvate molecules, but the only such interaction involving the molecules of 4 is shown in Fig. 2. This involves the platinum atom Pt(1) and one of the hydrogen atoms H(27b) of the tolyl group of a neighbouring molecule (related by the symmetry operation x, y, z + 1), at a distance of 2.8 Å.18 The effect is to link the molecules of 4 into chains that run through the crystal lattice in the c direction.
| Pt(1)–C(28) | 1.940(19) | Pt(1)–C(19) | 1.947(17) |
| Pt(1)–N(2) | 2.040(5) | Pt(1)–N(1) | 2.065(14) |
| N(1)–C(5) | 1.35(2) | N(2)–C(6) | 1.34(2) |
| C(5)–C(6) | 1.44(3) | C(19)–C(20) | 1.25(2) |
| C(20)–C(21) | 1.38(2) | C(28)–C(29) | 1.20(2) |
| C(29)–C(30) | 1.45(3) | ||
| C(28)–Pt(1)–C(19) | 91.7(7) | C(28)–Pt(1)–N(2) | 94.7(7) |
| C(19)–Pt(1)–N(2) | 173.6(6) | C(28)–Pt(1)–N(1) | 173.1(7) |
| C(19)–Pt(1)–N(1) | 95.0(6) | N(2)–Pt(1)–N(1) | 78.6(6) |
| C(6)–N(2)–Pt(1) | 114.7(13) | C(20)–C(19)–Pt(1) | 172.1(15) |
| N(2)–C(6)–C(5) | 117.1(17) | N(1)–C(5)–C(6) | 113.6(16) |
| C(19)–C(20)–C(21) | 175.0(18) | C(28)–C(29)–C(30) | 173(2) |
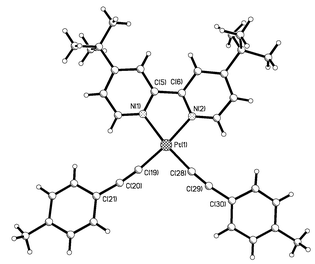 | ||
| Fig. 1 The crystal structure of compound 4. Three molecules of CHCl3 have been omitted for clarity. | ||
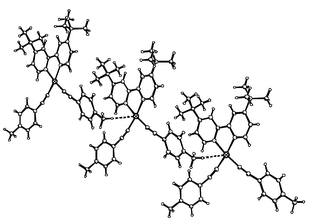 | ||
| Fig. 2 Part of the crystal structure of compound 4, showing the intermolecular interaction between H(27b) of one molecule and the platinum atom of a neighbouring molecule. | ||
In order to rationalise the differences in the spectra of compounds 2 and 3–5, some simple MO calculations based on the structures of 2 and 4 have been performed with the CACAO software package.19 The results show that for 4 the HOMO is largely metal based, with in-phase overlap of the dx2 − y2 orbital with the in-plane π-bonding combinations of the two acetylide triple bonds. (The compound is lying in the xy plane with the acetylide ligands pointing between these axes, in common with the axis system used in ref. 4.) The LUMO is approximately 91% bipyridyl based, in agreement with previous calculations.4 In 2, however, there are two degenerate LUMOs, each being based largely on the pz orbitals of one of the two nitro groups. The third lowest unoccupied MO is the first to be bipyridine based, but is only approximately 72% bipyridyl in nature and contains a large contribution from the nitro groups of the acetylide ligands.
Spectroelectrochemical behaviour
The electrochemical behaviour of compounds 2 and 3 is consistent with the results of the extended Hückel molecular orbital (EHMO) calculations. Cyclic voltammetry at 290 K reveals that both systems exhibit reversible reduction processes; complex 3 has a one-electron reversible reduction at −1.32 V, whereas 2 has a two-electron reversible reduction process at −1.08 V. Thus, it is easier to reduce the nitrated complex than the non-nitrated complex, and furthermore the dinitro complex is reduced in a single two-electron step (or in two very closely spaced one-electron steps that are not resolvable in cyclic voltammetry). Both complexes show reversible behaviour on an extended timescale, and were therefore subjected to spectroelectrochemical study.
In situ reduction of compound 3 to the radical monoanion [Pt(CCC6H5)2(tBu2bipy)]−3−− at −1.5 V at 240 K is shown in Fig. 3. The spectrum of the electrogenerated species shows a marked resemblance to that previously reported for [Pt(bipy)L2]−,4 and so the bands centred at 769, 500 and 370 nm are assigned to π → π* transitions of the co-ordinated bipyridyl anion, bipy−, and the band at 425 nm is assigned to a charge transfer transition. It may be concluded therefore that the one-electron reduction product 3−− should be formulated as [PtII(CCC6H5)2(tBu2bipy−)]. Note that there are no isosbestic points in the spectra of 3/3−− because the spectrum of the latter has a greater absorption coefficient at every wavenumber than that of the former. However, the spectrum of the parent species is completely regenerated if the electrogeneration potential is reset to 0 V after conversion into the anion is complete. The spectra obtained by reducing 2 under the same conditions are quite different from those obtained on reducing 3. The two absorptions at around 320 nm that are assigned as intraligand bipy π → π* transitions remain relatively unaffected, but the more intense band at 370 nm undergoes a considerable change (Fig. 4). None of the bands associated with the reduced bipyridyl ligand that are seen in Fig. 3 are generated, and so it may be inferred that the co-ordinated bipyridyl ligand is not the site of redox activity in this compound, and that the 370 nm absorption is not the same MLCT band seen for compounds 1 and 3–5.
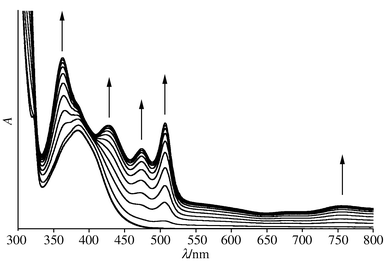 | ||
| Fig. 3 The spectral changes seen during the reduction of compound 3 to 3−−. | ||
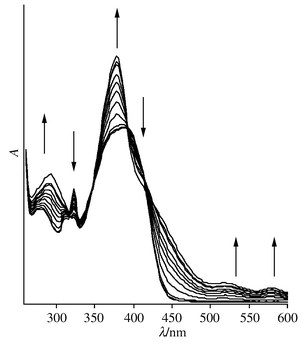 | ||
| Fig. 4 The spectral changes seen during the reduction of compound 2 to 2−−. | ||
The in situ generated solution EPR spectrum of compound 3−− is shown in Fig. 5, along with an excellent simulation. In the spectrum of the monoreduced anion hyperfine coupling of the unpaired electron to the platinum nucleus and superhyperfine coupling to the nuclei is observed, which may be modelled using the data given in Table 4. The spectrum shows a great similarity to the previously reported solution EPR spectrum of [Pt(bipy)(CN)2]−,4 and the coupling constants (also given in Table 4) are in remarkable agreement. Thus, the conclusions reached for [Pt(bipy)(CN)2]− must also apply in this system; that is, that the unpaired electron in 3−− resides on the co-ordinated tBu2bipy, in agreement with the predictions of the EHMO calculations and the UV/visible results given above. The simpler EPR spectrum for 3−− as opposed to the cyanide complex confirms that the protons in the 4 and 4′ positions in bipy couple to the unpaired electron in [Pt(bipy)(CN)2]−, since by substituting those positions with tert-butyl groups one set of superhyperfine coupling is removed. We therefore assign the 14N couplings in 3−− to the bipyridyl nitrogens, and the 1H couplings to one of the bipyridyl hydrogen atoms, though it is not possible to decide which. In addition, the similar EPR behaviours of 3−− and [Pt(bipy)(CN)2]− indicate that the phenylacetylide ligands are electronically analogous to cyanide ligands.
| 195Pt coupling | 14N coupling | 1H coupling | |||||
|---|---|---|---|---|---|---|---|
| Compound | No. | A iso/G | No. | A iso/G | No. | A iso/G | ΔHpp |
| 22−2− | 1 | 15.8 | 1 | 10.1 | 2 | 3.37 | 3.00 |
| 3−− | 1 | 21.0 | 2 | 3.37 | 2 | 2.85 | 3.2 |
| [Pt(bipy)(CN)2]−4 |
1 | 20.5 | 2 | 3.4 | 2 | 2.8 | |
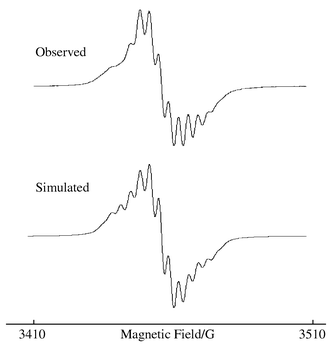 | ||
| Fig. 5 The observed and simulated EPR spectra of compound 3−−. | ||
The solution EPR spectrum of compound 22−2− and an excellent simulation of the same are shown in Fig. 6; the simulation parameters are also included in Table 4. The EPR spectra of 22−2− and 3−− are obviously very different, and hence require very different coupling constants to model them. The spectrum of 22−2− has significantly smaller coupling to the platinum nucleus and a very much larger coupling to 14N. Furthermore note that now the unpaired electron couples to only one 14N nucleus, whereas in 3−− it couples to two such nuclei. Thus we suggest that the site of redox activity in 2 is the nitrophenyl ligand, and more specifically the nitro group which carries much of the electron density when the compound is in the reduced state. It therefore has a degenerate pair of LUMOs based on the nitro groups which are orthogonal to each other and spatially well separated, meaning that the two electrons are not interacting with each other and the direduced complex can satisfactorily be modelled using an S = 1/2 system. The 1H coupling is presumably to two equivalent protons on the phenyl ring.
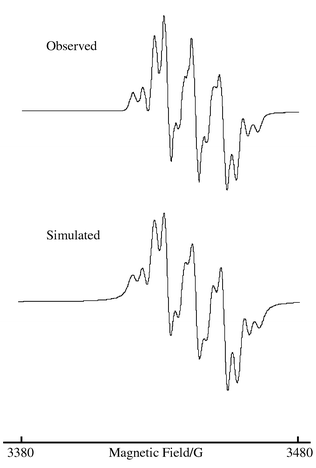 | ||
| Fig. 6 The observed and simulated EPR spectra of compound 22−2−. | ||
These findings indicate that the transition forming the 370 nm absorption band of compound 2 is based at least partly upon the nitro groups. The almost non-existent solvatochromism would seem to rule out this transition being charge transfer in nature, and therefore this band may tentatively be assigned as an intra-ligand feature.
Experimental
General
Solvents were pre-dried and distilled from appropriate drying agents.20 All chemicals were obtained from commercial sources and used as received, except for p-nitrophenylacetylene, which was prepared according to the literature procedure.21 The 1H NMR spectra were recorded on a Bruker AC 250 spectrometer and referenced to solvent resonances, IR spectra in dichloromethane in an NaCl cell on a Perkin-Elmer 1710 Fourier-transform spectrometer and +FAB mass spectra on a Kratos MS890 mass spectrometer. All electrochemical manipulations were performed under an inert atmosphere using a previously described experimental set-up,22 with platinum working and auxiliary electrodes and a Ag–AgCl reference electrode (against which the ferrocene–ferrocenium couple measures +0.55 V), in a 0.1 M solution of [nBu4N][BF4] in DMF at 250 mV s−1. UV/visible absorption spectra were obtained on a Perkin-Elmer λ9 spectrometer. The EPR spectra were recorded on a Bruker ER200-D X-band spectrometer, and simulations were performed using Bruker Simphonia. Microanalyses were carried out in the Department of Chemistry in Cambridge.Preparations
Compound 3 was a yellow solid obtained using phenylacetylene instead of TMSA, followed by recrystallisation from CH2Cl2–hexane. Yield: 0.156 g (0.234 mmol, 83%) (C34H34N2Pt.CH2Cl2 requires C, 56.06; H, 4.84; N, 3.74%; M 666. Found: C, 56.17; H, 4.80; N, 3.40%; M+, m/z 666.2). 1H NMR (CDCl3): δ 1.43 [s, 18 H, tBu], 7.1–7.3 [m, 6 H, phenyl CH], 7.5–7.6 [m, 6 H, phenyl CH and H5], 7.93 [d, 2 H, 4J(H3H5) = 1.8, H3] and 9.70 [d, 2 H, 3J(H5H6) = 5.3 Hz, H6].
Compound 4 was a yellow solid obtained using p-tolylacetylene instead of TMSA, followed by recrystallisation from CH2Cl2–hexane. Yield: 0.255 g (0.37 mmol, 69%) (C36H38N2Pt requires C, 62.30; H, 5.52; N, 4.04%; M 693. Found: C, 62.04; H, 5.57; N, 4.03%; M+, m/z 693.3). 1H NMR (CDCl3): δ 1.43 [s, 18 H, tBu], 2.31 [s, 6 H, tolyl CH3], 7.04 [d, 4 H, 3J(HH) = 8, tolyl CH], 7.42 [d, 4 H, tolyl CH], 7.54 [dd, 2 H, 3J(H5H6) = 5.9, 4J(H3H5) = 1.8 Hz, H5], 7.92 [d, 2 H, H3] and 9.72 [d, 2 H, H6].
Crystallography
Yellow crystals of compound 4·3CHCl3 were obtained by slow evaporation of a chloroform solution. Suitable crystals were mounted using a perfluoroether oil, and cooled to 153 K with an Oxford Cryostream apparatus. Data were collected on a Rigaku AFC7R diffractometer using the ω–2θ technique. Semiempirical absorption corrections based upon ψ scans were applied (TEXSAN).24 The structure was solved by Patterson methods (SHELXS 86)25 and subsequent Fourier-difference syntheses, and refined by full-matrix least squares on F2 (SHELXL 97).26 The platinum and chlorine atoms were refined with anisotropic displacement factors, and all aromatic hydrogen atoms were placed in idealised positions and allowed to ride on the relevant carbon atoms. Methyl groups were given an idealised tetrahedral geometry and allowed to rotate during the refinement. A final electron density difference map showed no regions of significant electron density.
1 (no. 2), a = 11.115(3), b = 17.187(2), c = 10.573(4) Å, α = 103.58(2), β = 93.90(2), γ = 75.67(2)°, U = 2123.5(9) Å3, T = 290(2) K, Z = 2, μ(Mo-Kα) = 3.901 mm−1, 4607 reflections measured, 4382 unique (Rint = 0.1580) which were used in all calculations. The final residuals on 264 parameters were R1 = 0.068 and wR2 = 0.195 for 3539 reflections with I > 2σ(I), and R1 = 0.097, wR2 = 0.226 for all data.
CCDC reference number 186/1736.
See http://www.rsc.org/suppdata/dt/a9/a907028a/ for crystallographic files in .cif format.
Acknowledgements
We would like to thank the EPSRC for financial support (C. J. A., P. R. R. and S. L. J.), and the University of Edinburgh and the committee of vice-chancellors and principals for an overseas research studentship (X. L.). We are grateful to Johnson Matthey for the generous loan of platinum salts.References
- J. Forniés and E. Lalinde, J. Chem Soc., Dalton Trans., 1996, 2587 RSC; M. D. Janssen, K. Köhler, M. Herres, A. Dedieu, W. J. J. Smeets, A. L. Spek, D. M. Grove, H. Lang and G. van Koten, J. Am. Chem. Soc., 1996, 118, 4817 CrossRef CAS; A. Kovács and G. Frenking, Organometallics, 1999, 18, 887 CrossRef CAS; M. D. Janssen, M. Herres, L. Zsolnai, A. L. Spek, H. Lang and G. van Koten, Organometallics, 1995, 14, 1098 CrossRef CAS; S. Lotz, P. H. van Rooyen and R. Meyer, Adv. Organomet. Chem., 1995, 37, 219 Search PubMed; S. Yamazaki and A. J. Deeming, J. Chem. Soc., Dalton Trans., 1993, 3051 RSC; Y. Hayishi, M. Osawa, K. Kobayashi, T. Sato, M. Sato and Y. Wakatsuki, J. Organomet. Chem., 1998, 569, 169 CrossRef; H. Lang and G. Rheinwald, J. Prakt. Chem., 1999, 341, 1 CrossRef CAS.
- C. J. Adams and P. R. Raithby, J. Organomet. Chem., 1999, 578, 178 CrossRef CAS.
- R. H. Herber, M. Croft, M. J. Coger, B. Bilash and A. Sakiner, Inorg. Chem., 1994, 33, 2422 CrossRef CAS; W. B. Connick, R. E. Marsh, W. B. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913 CrossRef CAS; W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6261 CrossRef CAS; W. B. Connick, L. M. Henling and R. E. Marsh, Acta Crystallogr., Sect. B, 1996, 52, 817 CrossRef; M. Kato, C. Kosuge, S. Yano and M. Kimura, Acta Crystallogr., Sect. C, 1997, 53, 838 CrossRef.
- D. Collison, F. E. Mabbs, E. J. L. McInnes, K. J. Taylor, A. J. Welch and L. J. Yellowlees, J. Chem. Soc., Dalton Trans., 1996, 329 RSC; E. J. L. McInnes, R. D. Farley, K. J. Taylor, L. J. Yellowlees and C. C. Rowlands, J. Chem. Soc., Faraday Trans., 1998, 2985 RSC.
- L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735 CrossRef CAS.
- K. C. Shih and R. H. Herber, Inorg. Chem., 1992, 31, 5444 CrossRef CAS.
- S. L. James, M. Younus, P. R. Raithby and J. Lewis, J. Organomet. Chem., 1997, 543, 237 CrossRef CAS.
- Y. Y. Ng, C. M. Che and S. M. Peng, New J. Chem., 1996, 20, 781 Search PubMed; C. W. Chan, L. K. Cheng and C. M. Che, Coord. Chem. Rev., 1994, 132, 87 CrossRef CAS.
- C. J. Adams, S. L. James and P. R. Raithby, Chem. Commun., 1997, 2155 RSC; J. Terheijden, G. Van Koten, F. Muller, D. M. Grove, K. Vrieze, E. Nielson and C. H. Stam, J. Organomet. Chem., 1986, 315, 401 CrossRef CAS.
- J. Geisenberger, U. Nagel, A. Sebald and W. Beck, Chem. Ber., 1983, 116, 911 Search PubMed.
- S. Achar, J. D. Scott, J. J. Vittal and R. J. Puddephatt, Organometallics, 1992, 12, 4592.
- C. K. McGill , US Pat., 4 267 335, 1981. Search PubMed.
- G. T. Morgan and F. H. Burstall, J. Chem. Soc., 1934, 965 RSC.
- F. R. Hartley, S. G. Murray and C. P. McAuliffe, Inorg. Chem., 1979, 18, 1394 CrossRef CAS.
- R. Nast and R. Kramolowsky, Chem. Ber., 1975, 108, 1511 Search PubMed.
- P. M. Gidney, R. D. Gillard and B. T. Heaton, J. Chem. Soc., Dalton Trans., 1973, 133 Search PubMed.
- J. Manna, K. D. John and M. D. Hopkins, Adv. Organomet. Chem., 1995, 38, 79 Search PubMed.
- A. Martin, J. Chem. Educ., 1999, 76, 578 Search PubMed.
- C. Mealli and J. Proserpio , CACAO, PC version 4, ISSECC, Florence, 1994 Search PubMed; J. Chem. Educ., 1990, 67, 399. Search PubMed.
- W. L. F. Armargo and D. D. Perrin , Purification of Laboratory Chemicals, 4th edn., Butterworth-Heinemann, Oxford, 1996. Search PubMed.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 CrossRef CAS.
- S. A Macgregor , E. J. L. McInnes , R. J. Sorbie and L. J. Yellowlees , Molecular Electrochemistry of Inorganic, Bioorganic and Organometallic Compounds, eds. A. J. L. Pombeiro and J. A. McCleverty, Kluwer, Dordrecht, 1993; Search PubMed; E. J. L. McInnes, R. D. Farley, C. C. Rowlands, A. J. Welch, L. Rovatti and L. J. Yellowlees, J. Chem. Soc., Dalton Trans., 1999, 4203 Search PubMed.
- P. Belser and A. Von Zelewsky, Helv. Chim. Acta, 1980, 63, 1675 CrossRef CAS.
- TEXSAN, Single Crystal Structure Analysis Software, Version 1.7, Molecular Structure Corporation, The Woodlands, TX, 1995. Search PubMed.
- G. M. Sheldrick , SHELXS 86, University of Göttingen, 1986. Search PubMed.
- G. M. Sheldrick , SHELXL 97, University of Göttingen, 1997. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |