Synthesis of ruthenium complexes with planar-chiral cyclopentadienyl-pyridine or -phosphine bidentate ligands
Noriko
Dodo
,
Yuji
Matsushima
,
Mitsunari
Uno
,
Kiyotaka
Onitsuka
and
Shigetoshi
Takahashi
*
The Institute of Scientific and Industrial Research, Osaka University, Mihogaoka 8-1, Ibaraki, Osaka, 567-0047, Japan
First published on 24th December 1999
Abstract
Novel ruthenium complexes possessing planar-chiral cyclopentadienyl-pyridine or -phosphine bidentate ligands have been synthesized from (trisubstituted cyclopentadienyl)(benzene)ruthenium complexes. Co-ordination of the pyridyl group linked to a cyclopentadienyl ring through an ester bond is fairly weak, so that the pyridine dissociates from the central ruthenium atom in acetonitrile. In contrast, the phosphine ligand tightly co-ordinates to the ruthenium even in acetonitrile. Enantiomerically pure samples of planar-chiral cyclopentadienylphosphine complexes have been prepared by use of optically pure ruthenium complexes as a starting material. The molecular structure of a neutral ruthenium dithiocarbamate complex with the cyclopentadienylphosphine bidentate ligand has been determined by X-ray crystallography.
Introduction
It is widely accepted that optically active organometallic complexes are powerful tools for asymmetric organic syntheses as effective catalytic or stoichiometric mediators.1 Half-sandwich complexes MCpL3, which are referred to as “three-legged piano stools”, have a rich chemistry and chiral versions have been applied to asymmetric catalysis.2 There are several examples of asymmetric reactions catalysed by half-sandwich complexes with chiral ligands.3 Although some optically active complexes with a chiral metal center have also been prepared, application of these complexes is limited to stoichiometric reactions since racemization at the chiral metal center often takes place during the reactions.4 Planar-chiral cyclopentadienyl metal complexes should be useful as asymmetric catalysts because racemization arising from a change in co-ordination mode from η5 into η1 or dissociation of the cyclopentadienyl group is unlikely to occur. The efficiency of planar-chiral cyclopentadienyl complexes has proved particularly valuable in the polymerization of olefins by bis(cyclopentadienyl) complexes (metallocenes) of Group IV metals.5 On the other hand, few studies on planar-chiral cyclopentadienyl complexes of late transition metals have so far been made though they have great potential as catalysts in novel asymmetric reactions.6 Recently Fu and co-workers![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 7 described asymmetric reactions using planar-chiral ferrocene derivatives as catalysts,7 in which, however, metal atoms with planar-chiral ligands do not act as a reactive site.
7 described asymmetric reactions using planar-chiral ferrocene derivatives as catalysts,7 in which, however, metal atoms with planar-chiral ligands do not act as a reactive site.
Previously we have reported the syntheses of planar-chiral organometallic complexes of late transition metals with trisubstituted η5-cyclopentadienyl ligands.8,9 As a fundamental investigation of the application of planar-chiral complexes to asymmetric catalysts, we have examined some stoichiometric reactions of these planar-chiral complexes. The stereoselectivities induced by these planar chiral complexes, however, are not high. One possible reason may be rotation of the cyclopentadienyl groups, which could cause corruption of the asymmetric environment around a reactive metal center. In order to prevent this we set out to tether the cyclopentadienyl group to anchor ligands.10 Herein we report the synthesis of enantiomerically pure planar-chiral cyclopentadienyl ruthenium complexes possessing such anchor ligands. Although a few planar-chiral complexes with cyclopentadienyl- and indenyl-phosphine bidentate ligands have been prepared as racemic mixtures,6 this is the first example of an enantiomerically pure one. Very recently planar-chiral η6-arene complexes with anchor ligands have been isolated in an optically pure state by Ward and co-workers.11
Results and discussion
First we chose pyridine as an anchor ligand and attempted to synthesize cyclopentadienylpyridine ruthenium complexes. Cationic ruthenium complexes [Ru{η5-C5H2(Me)(R)CO2Et}(η6-C6H6)][PF6] (R = Me 1a, Ph 1b or t-Bu 1c) that possess a trisubstituted cyclopentadienyl ligand with an exchangeable ester group were used as starting materials.9 Hydrolysis of 1a–1c under basic conditions gave carboxylic acid analogs [Ru{η5-C5H2(Me)(R)CO2H}(η6-C6H6)][PF6] (R = Me 2a, Ph 2b or t-Bu 2c) (Scheme 1). A 2-pyridyl group was introduced by esterification with 2-NC5H4CH2OH using a standard method to give [Ru{η5-C5H2(Me)(R)CO2CH2C5H4N}(η6-C6H6)][PF6] (R = Me 3a or Ph 3b). Irradiation with UV light in CH3CN caused dissociation of the η6-benzene ligand to give [Ru{η5,η1-C5H2(Me)(R)CO2CH2C5H4N}(CH3CN)2][PF6] (R = Me 4a or Ph 4b) in quantitative yields.12 The elemental analysis showed that the proposed structure is consistent with the composition of 4a. However, complexes 4a and 4b are unstable in common organic solvents other than acetonitrile even at low temperature. In their 1H NMR spectra in acetonitrile-d3, chemical shifts of the signals attributed to pyridyl protons were very similar to those of 3a and 3b, respectively. These data suggest that the pyridyl groups of 4a and 4b dissociate from the ruthenium atom to generate [Ru{η5-C5H2(Me)(R)CO2CH2C5H4N}(CH3CN)3][PF6] (R = Me 5a or Ph 5b) in acetonitrile.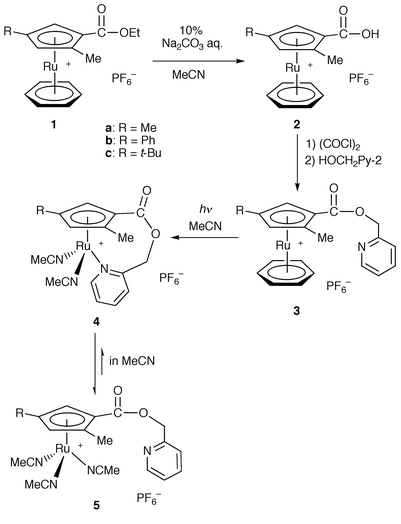 | ||
| Scheme 1 | ||
On treatment of complexes 4a and 4b with bipyridine in acetonitrile, two acetonitrile ligands were replaced by 2,2′-bipyridine to give [Ru{η5,η1-C5H2(Me)(R)CO2CH2C5H4N}(bipy)][PF6] (R = Me 6a or Ph 6b) (Scheme 2) which are stable not only in acetonitrile but also in acetone at room temperature. The 1H NMR spectra of 6a, which were measured both in acetonitrile-d3 and in acetone-d6, are shown in Fig. 1 along with that of 3a in acetone-d6. The signals assigned to α- and γ-protons of the anchor pyridyl group in 3a were observed at δ 8.66 and 7.87, respectively. There is no significant difference in these chemical shifts between acetonitrile-d3 and acetone-d6. The 1H NMR spectrum of 6a in acetonitrile-d3 showed signals due to the pyridyl protons of the anchor ligand at δ 8.54 and 7.72, which are close to those of 3a. In contrast, the signals attributed to the pyridyl protons at α- and γ-positions of 6a were observed at δ 9.14 and 8.02 in acetone-d6, respectively. These data indicate that the pyridyl group of 6a dissociates from the ruthenium atom in acetonitrile to form an acetonitrile complex, [Ru{η5-C5H2(Me)(R)CO2CH2C5H4N}(bipy)(CH3CN)][PF6] (R = Me 7a or Ph 7b), but co-ordinates in acetone. The signals of methylene protons on the tether were observed as two diastereotopic doublets due to the planar chirality of the cyclopentadienyl group. It may be of interest that the difference in the chemical shift between the two diastereotopic methylene protons of 6a in acetone is much larger than that in acetonitrile. Two methylene signals were observed at δ 5.72 and 4.55 in acetone-d6 whereas δ equals 5.28 and 5.21 in acetonitrile-d3, respectively. The co-ordination of the pyridyl group would amplify the magnetic inequivalency of the geminal protons.13
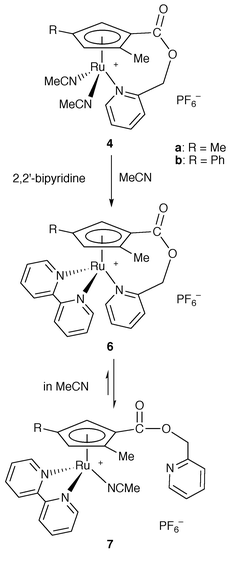 | ||
| Scheme 2 | ||
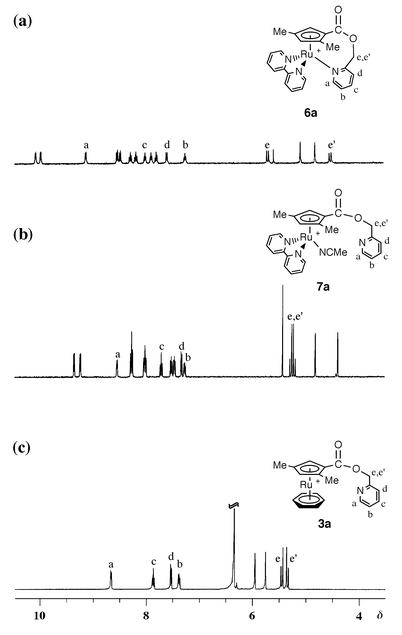 | ||
| Fig. 1 The 1H NMR spectra of cyclopentadienylpyridine ruthenium complexes: (a) 6a in acetone-d6, (b) 6a in acetonitrile-d3 generating 7a and (c) 3a in acetone-d6. | ||
Since co-ordination of the anchor pyridyl group seems to be weak, it was changed to a diphenylphosphino group, which was similarly introduced by esterification of complexes 2a–2c with HO(CH2)nPPh2.14 However, the co-ordination of η6-benzene is so strong that the phosphorus atom does not co-ordinate to the ruthenium atom in [Ru{η5-C5H2(Me)(R)CO2(CH2)nPPh2}(η6-C6H6)][PF6] (R = Me, n = 2 8a; R = Ph, n = 2 8b; R = t-Bu, n = 2 8c; R = Me, n = 3 8d) (Scheme 3). Irradiation by UV light in CH3CN caused dissociation of the η6-benzene ligand to give [Ru{η5,η1-C5H2(Me)(R)CO2(CH2)nPPh2}(CH3CN)2][PF6] (R = Me, n = 2 9a; R = Ph, n = 2 9b; R = t-Bu, n = 2 9c; R = Me, n = 3 9d) in quantitative yields.12 Complexes 9a–9d are stable in air and soluble in polar solvents such as dichloromethane, chloroform and acetone.
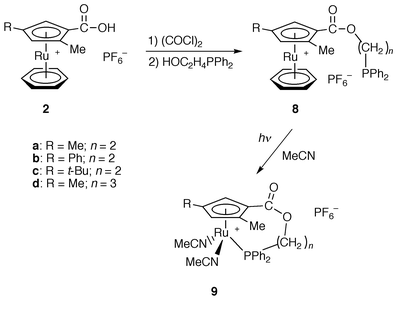 | ||
| Scheme 3 | ||
Complexes 9a–9d were fully characterized by means of spectral analyses. The FAB mass spectrum of 9a revealed a peak at m/z = 533, which corresponds to the cationic species containing Ru, suggesting that 9a has two CH3CN ligands as well as a cyclopentadienylphosphine ligand in the molecule. The result of elemental analysis also supported the composition of 9a. The 31P NMR of 9a showed a singlet at δ 35.56, while the signal of complex 8a was observed at much higher magnetic field (δ −20.89). The remarkable difference in chemical shifts of the 31P NMR spectra between 9a and 8a indicates the co-ordination of an anchor PPh2 group linked by the tether to a cyclopentadienyl ligand.15 The 1H NMR spectrum of 9a in CDCl3 exhibited four multiplet signals assignable to the protons of two methylene groups on the tether as observed for 6a in acetone-d6. The signals attributable to the protons of two CH3CN ligands appeared as two doublets due to coupling with the phosphorus nucleus. Similarly in the 13C NMR spectrum of 9a, coupling with the phosphorus atom was also found for the signals due to cyclopentadienyl carbon at the 3 position. These data clearly identified the structure of 9a as involving co-ordination of a cyclopentadienylphosphine “bidentate” ligand to the central ruthenium atom. It should be noted that no significant changes have been observed in the 1H and 31P NMR spectra of 9a in CD3CN relative to those in acetone-d6, suggesting that the phosphino group, in contrast to the pyridyl group, is tightly bound to the ruthenium atom.
Previously we have reported that an enantiomerically pure sample of the planar-chiral cyclopentadienyl complex (S)-[Ru{η5-C5H2Me2CO2C10H19-(L)}(η6-C6H6)][PF6] (S)-1d is obtained by fractional recrystallization.9 Enantiomer (R)-1d is also obtained as a (D)-menthyl ester analog. Thus, both complexes (R)- and (S)-1d were successfully converted into cyclopentadienylphosphine complexes (R)- and (S)-9a by similar reaction to that of racemic 9a (Scheme 4). A 1H NMR experiment using a chiral shift reagent, [Eu(hfc)3] [hfc = 3-(heptafluoropropylhydroxymethylene)-D-camphorate], clearly showed that no racemization of the planar-chiral cyclopentadienyl ligand occurred during the conversion from (R)- and (S)-1d into (R)- and (S)-9a. Therefore, (R)- and (S)-9a showed the same melting points, the same absolute values but with opposite sign in the specific rotations [α]D, and mirror image circular dichroism (CD) spectra in all regions, as depicted in Fig. 2. These results unequivocally suggest that complexes (R)- and (S)-9a are a pair of enantiomers in an optically pure form.
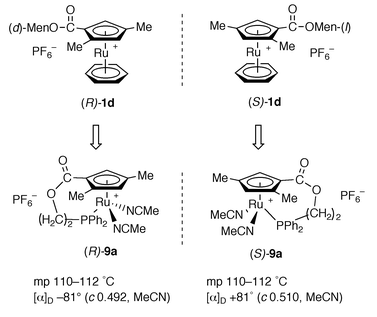 | ||
| Scheme 4 | ||
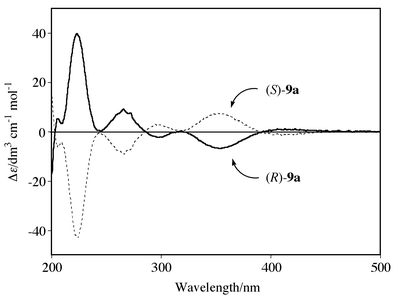 | ||
| Fig. 2 The CD spectra of complexes (R)- and (S)-9a in acetonitrile. | ||
In order to obtain the structural information on the cyclopentadienylphosphine bidentate ligand, we attempted X-ray crystallography of complexes 9a–9d. However, single crystals did not grow upon recrystallization. Thus we converted the cationic complex into a neutral complex with expectation of good crystallinity. Treatment of 9b with sodium dithiocarbamate in refluxing dichloromethane gave a dithiocarbamate complex, [Ru{η5,η1-C5H2(Me)(Ph)CO2(CH2)2PPh2}(S2CNMe2)] 10, in 40% yield (Scheme 5).16 As was expected, good crystals of 10 were obtained by recrystallization from dichloromethane–diethyl ether. The X-ray crystallographic analysis unequivocally showed that 10 has a three-legged piano stool structure with a cyclopentadienylphosphine bidentate ligand. The molecular structure is illustrated in Fig. 3, and selected bond distances and angles are given in Table 1. The structural parameters around the ruthenium atom are similar to those of the non-chelated complex [Ru(η5-C5H5)(PPh3)(S2NMe2)].16,17 The phenyl ring C(7)–C(12) and carbonyl planes defined by C(1), C(13), O(1) and O(2) are slightly twisted relative to the cyclopentadienyl ring, and the torsion angles are 15.8 and 25.8°, respectively. The most striking structural feature of 10 is the conformation of the two phenyl groups on the phosphorus atom. One phenyl group C(22)–C(27) is oriented axially with respect to the plane defined by S(1), S(2) and P(1) and is accommodated on the side of the dithiocarbamate group to minimize the steric repulsion. In contrast, the other phenyl group C(16)–C(21) protrudes equatorially. The phenyl group C(7)–C(12) on the cyclopentadienyl ring also exerts steric influence on substrates interacting with the ruthenium atom. Therefore, the dithiocarbamate ligand on the ruthenium atom lies in a chiral environment constructed by substituents on the cyclopentadienyl ring and phenyl groups on the phosphorus atom, suggesting that the ruthenium complexes with the trisubstituted cyclopentadienylphosphine bidentate ligands have a great potential applicable to stoichiometric or catalytic asymmetric reactions.
| Ru(1)–S(1) | 2.3928(7) | Ru(1)–S(2) | 2.4022(9) |
| Ru(1)–P(1) | 2.3100(7) | Ru(1)–C(1) | 2.163(3) |
| Ru(1)–C(2) | 2.219(3) | Ru(1)–C(3) | 2.233(3) |
| Ru(1)–C(4) | 2.214(3) | Ru(1)–C(5) | 2.170(3) |
| S(1)–C(28) | 1.709(3) | S(2)–C(28) | 1.725(3) |
| N(1)–C(28) | 1.320(4) | N(1)–C(29) | 1.450(4) |
| N(1)–C(30) | 1.460(5) | ||
| S(1)–Ru(1)–S(2) | 72.02(3) | S(1)–Ru(1)–P(1) | 90.92(3) |
| S(1)–Ru(1)–C(1) | 164.99(8) | S(1)–Ru(1)–C(2) | 133.42(8) |
| S(1)–Ru(1)–C(3) | 103.12(8) | S(1)–Ru(1)–C(4) | 101.36(8) |
| S(1)–Ru(1)–C(5) | 130.64(8) | S(2)–Ru(1)–P(1) | 88.20(3) |
| S(2)–Ru(1)–C(1) | 117.44(8) | S(2)–Ru(1)–C(2) | 96.36(8) |
| S(2)–Ru(1)–C(3) | 109.76(8) | S(2)–Ru(1)–C(4) | 146.31(8) |
| S(2)–Ru(1)–C(5) | 156.47(8) | P(1)–Ru(1)–C(1) | 100.69(8) |
| P(1)–Ru(1)–C(2) | 134.64(8) | P(1)–Ru(1)–C(3) | 159.82(8) |
| P(1)–Ru(1)–C(4) | 125.35(8) | P(1)–Ru(1)–C(5) | 96.64(8) |
| Ru(1)–S(1)–C(28) | 89.0(1) | Ru(1)–S(2)–C(28) | 88.33(9) |
| C(28)–N(1)–C(29) | 122.1(3) | C(28)–N(1)–C(30) | 120.8(3) |
| C(29)–N(1)–C(30) | 117.0(3) | S(1)–C(28)–S(2) | 110.3(2) |
| S(1)–C(28)–N(1) | 125.3(2) | S(2)–C(28)–N(1) | 124.3(2) |
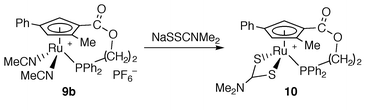 | ||
| Scheme 5 | ||
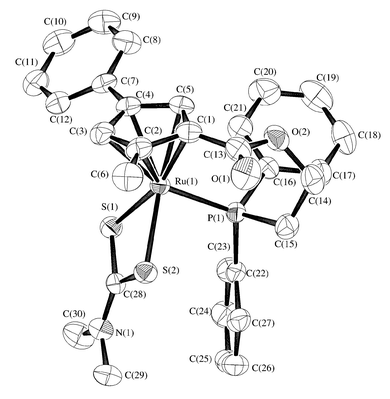 | ||
| Fig. 3 Molecular structure of complex 10. Hydrogen atoms are omitted for clarity. | ||
Conclusion
We have developed a convenient synthetic route to planar-chiral cyclopentadienyl-pyridine or -phosphine ruthenium complexes. The phosphino group tightly co-ordinates to the ruthenium atom whereas the pyridyl group dissociates in acetonitrile. The cyclopentadienylphosphine ligand provides a good chiral environment around the ruthenium atom as a result of the steric effect of substituents on the cyclopentadienyl ring and phenyl groups of the phosphine ligand. We are currently investigating application of these complexes as catalysts for asymmetric organic reactions.Experimental
General considerations
All reactions were carried out under an atmosphere of argon, but the work-up was performed in air. NMR Spectra were measured in CDCl3 on JEOL JNM-LA400, JNM-LA600 and Bruker ARX400 spectrometers. In 1H and 13C NMR, SiMe4 was used as an internal standard, and an external 85% H3PO4 reference was used for 31P NMR. FAB Mass spectra were obtained on a JMS-600H instrument, and IR spectra on a Perkin-Elmer System 2000 FT-IR. Optical rotations and CD spectra were measured on JASCO DIP-1000 and JASCO J-725 instruments, respectively. Elemental analyses were performed by The Material Analysis Center, ISIR, Osaka University. The starting ruthenium complexes [Ru{η5-C5H2(Me)(R)CO2R′}(η6-C6H6)][PF6] (R = Me, R′ = Et 1a; R = Ph, R′ = Et 1b; R = t-Bu, R′ = Et 1c; R = Me, R′ = (L)- or (D)-menthyl 1d) were prepared by a method previously reported.9 2-(Diphenylphosphino)ethanol HO(CH2)2PPh2 was prepared according to the literature method,14 and 3-(diphenylphosphino)propanol HO(CH2)3PPh2 by a similar method using HO(CH2)3Cl in place of HO(CH2)2Cl.Preparations
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1]: 1732 (CO). 1H NMR (CDCl3): δ 8.63 (d, 1 H, J = 4.9), 7.82 (dd, 1 H, J = 7.9, 6.9), 7.45 (d, 1 H, J = 7.9), 7.33 (dd, 1 H, J = 4.9, 6.9), 6.08 (s, 6 H), 5.79 (s, 1 H), 5.66 (s, 1 H), 5.45 (d, 1 H, J = 12.9), 5.30 (d, 1 H, J = 12.9 Hz), 2.28 (s, 3 H) and 2.06 (s, 3 H). FAB MS: m/z 468 (M+ − PF6). Calc. for C20H20F6NO2PRu: C, 43.49; H, 3.65; F, 20.63; N, 2.54; P, 5.61. Found: C, 43.32; H, 3.57; F, 20.58; N, 2.55; P, 5.58%.
/cm−1]: 1737 (CO). 1H NMR (CDCl3): δ 8.68 (d, 1 H, J = 1.5), 7.89 (m, 1 H), 7.72 (m, 2 H), 7.58 (d, 1 H, J = 7.9), 7.41 (m, 4 H), 6.57 (s, 1 H), 6.40 (s, 1 H), 6.29 (s, 6 H), 5.52 (d, 1 H, J = 13.5), 5.40 (d, 1 H, J = 13.5 Hz) and 2.43 (s, 3 H). FAB MS: m/z 170 (M+ − PF6). Calc. for C25H22F6NO2PRu: C, 48.87; H, 3.65; F, 18.55; N, 2.28; P, 5.04. Found: C, 48.92; H, 3.41; F, 18.72; N, 2.21; P, 5.10%.
/cm−1]: 1714 (CO). 1H NMR (CD3CN): δ 8.53 (d, 1 H, J = 5.1), 7.66 (dd, 1 H, J = 7.5, 7.4), 7.41 (d, 1 H, J = 7.5), 7.27 (t, 1 H, J = 7.4, 5.1), 5.28 (d, 1 H, J = 1.3), 5.22 (d, 1 H, J = 1.3 Hz), 4.60 (s, 1 H), 4.10 (s, 1 H), 1.95 (s, 3 H) and 1.72 (s, 3 H). Calc. for C18H20F6N3O2PRu: C, 38.86; H, 3.62; F, 20.49; N, 7.55; P, 5.57. Found: C, 38.97; H, 3.64; F, 20.90; N, 7.98; P, 5.38%.
/cm−1]: 1715 (CO). 1H NMR (CD3CN): δ 8.55 (d, 1 H, J = 4.9 Hz), 7.79 (m, 1 H), 7.47 (m, 3 H), 7.33 (m, 4 H), 7.29 (m, 4 H), 5.46 (s, 1 H), 4.79 (s, 1 H), 3.61 (m, 2 H) and 2.06 (s, 3 H). An analytically pure sample was not obtained due to low thermal stability of 4b.
/cm−1]: 1694 (CO). 1H NMR (acetone-d6): δ 10.10 (d, 1 H, J = 5.4), 10.00 (d, 1 H, J = 6.1), 9.14 (d, 1 H, J = 5.1), 8.55 (d, 1 H, J = 7.8), 8.49 (d, 1 H, J = 8.2), 8.30 (dd, 1 H, J = 7.8, 6.8), 8.20 (dd, 1 H, J = 7.2, 6.8), 8.02 (dd, 1 H, J = 7.1, 6.7), 7.91 (dd, 1 H, J = 7.3, 6.7), 7.82 (dd, 1 H, J = 7.8, 6.6), 7.61 (d, 1 H, J = 8.1), 7.27 (dd, 1 H, J = 6.2, 6.3), 5.72 (d, 1 H, J = 14.5), 5.11 (s, 1 H), 4.84 (s, 1 H), 4.55 (d, 1 H, J = 14.5 Hz), 1.64 (s, 3 H) and 1.48 (s, 3 H). FAB MS: m/z 485 (M+ − PF6). Calc. for C24H22F6N3O2PRu: C, 45.72; H, 3.52; F, 18.08; N, 6.66; P, 4.91. Found: C, 45.56; H, 3.28; F, 18.00; N, 6.57; P, 4.90%.
/cm−1]: 1721 (CO). 1H NMR (CDCl3): δ 7.48–7.43 (m, 4 H), 7.38–7.26 (m, 6 H), 6.08 (s, 6 H), 5.64 (s, 1 H), 5.41 (s, 1 H), 4.47–4.40 (m, 2 H), 2.56–2.50 (m, 2 H), 2.29 (d, 3 H, J = 2.2) and 2.25 (d, 3 H, J = 2.2 Hz). 31P NMR (CDCl3): δ
−20.89 (s) and −142.67 (sept, J = 712 Hz). FAB MS: m/z 529 (M+ − PF6). Calc. for C28H28F6O2P2Ru: C, 49.93; H, 4.19; F, 16.92; P, 9.20. Found: C, 49.92; H, 4.08; F, 16.73; P, 9.36%.
/cm−1]: 1724 (CO). 1H NMR (CDCl3): δ 7.52–7.27 (m, 15 H), 6.26 (s, 1 H), 5.98 (s, 1 H), 5.97 (s, 6 H), 4.54–4.48 (m, 2 H), 2.66–2.50 (m, 2 H) and 2.37 (s, 3 H). 31P NMR (CDCl3): δ
−17.10 (s) and −138.99 (sept, JP-F = 708 Hz). FAB MS: m/z 591 (M+ − PF6). Calc. for C25H22F6NO2PRu: C, 48.87; H, 3.65; F, 18.55; N, 2.28; P, 5.04. Found: C, 48.92; H, 3.41; F, 18.72; N, 2.21; P, 5.10%.
/cm−1]: 1715 (CO). 1H NMR (acetone-d6): δ 7.60–7.49 (m, 4 H), 7.40 (d, 6 H, J = 8.3 Hz), 6.42 (s, 6 H), 5.75 (s, 1 H), 5.68 (s, 1 H), 4.49–4.43 (m, 2 H), 2.70–2.60 (m, 2 H), 2.30 (s, 3 H) and 1.20 (s, 9 H). 31P NMR (acetone-d6): δ
−17.27 (s) and −138.99 (sept, JP-F = 708 Hz). FAB MS: m/z 572 (M+ − PF6). Calc. for C31H34F6O2P2Ru: C, 52.03; H, 4.79; F, 15.93; P, 8.66. Found: C, 51.77; H, 4.75; F, 16.00; P, 8.77%.
/cm−1]: 1732 (CO). 1H NMR (CDCl3): δ 7.45–7.34 (m, 10 H), 6.03 (s, 6 H), 5.68 (s, 1 H), 5.65 (s, 1 H), 4.35–4.32 (m, 2 H), 2.25 (s, 3 H), 2.15–2.04 (m, 2 H), 2.04 (s, 3 H) and 1.85 (m, 2 H). 31P NMR (acetone-d6): δ
−11.78 (s) and −144.35 (sept, JP-F = 712 Hz). FAB MS: m/z 543 (M+ − PF6). Calc. for C29H30F6O2P2Ru: C, 50.66; H, 4.40; F, 16.58; P, 9.01. Found: C, 50.40; H, 4.21; F, 16.50; P, 9.22%.
/cm−1]: 1715 (CO). 1H NMR (CDCl3): δ 7.58–7.45 (m, 5 H), 7.31–7.25 (m, 3 H), 6.90 (t, 2 H, J = 8.1), 5.14 (s, 1 H), 5.06–4.97 (m, 1 H), 4.22 (s, 1 H), 3.97–3.90 (m, 1 H), 2.83–2.75 (m, 1 H), 2.57–2.50 (m, 1 H), 2.16 (d, 3 H, JP-H = 1.0), 2.11 (d, 3 H, JP-H = 1.2 Hz), 2.01 (s, 3 H) and 1.84 (s, 3 H). 13C NMR (acetone-d6): δ 166.52 (s), 135.06 (d, JP-C = 38.2), 133.95 (d, JP-C = 12.4), 131.20 (d, JP-C = 2.4), 130.41 (d, JP-C = 8.1), 130.08 (d, JP-C = 42.2), 129.20 (d, JP-C = 1.9), 128.96 (d, JP-C = 10.4), 127.85 (d, JP-C = 12.4), 126.89 (s), 125.61 (s), 108.17 (s), 98.41 (s), 77.79 (d, JP-C = 12.4), 74.47 (s), 68.81 (s), 59.21 (d, JP-C = 3.7), 17.72 (d, JP-C = 30.5 Hz), 11.70 (s), 11.22 (s), 2.82 (s) and 2.60 (s). 31P NMR (CDCl3): δ 35.56 (s) and −142.92 (sept, JP-F = 712 Hz). FAB MS: m/z 533 (M+ − PF6). Calc. for C28H28F6N2O2P2Ru: C, 46.09; H, 4.17; F, 16.32; N, 4.16; P, 9.14. Found: C, 45.87; H, 3.91; F, 16.66; N, 4.30; P, 9.00%.
/cm−1]: 1727 (CO). 1H NMR (CDCl3): δ 7.94–7.70 (m, 2 H), 7.56–7.48 (m, 3 H), 7.42–7.35 (m, 3 H), 7.06 (t, 2 H, J = 8.1), 5.18 (d, 1 H, J = 1.7), 5.05–4.96 (m, 1 H), 4.46 (d, 1 H, J = 1.7), 3.93–3.87 (m, 1 H), 2.89–2.81 (m, 1 H), 2.57–2.50 (m, 1 H), 2.18 (d, 3 H, JP-H = 1.0), 2.16 (d, 3 H, JP-H = 1.5 Hz), 2.06 (s, 3 H) and 1.26 (s, 9 H). 13C NMR (acetone-d6): δ 167.61 (s), 134.42 (d, JP-C = 38.9), 134.13 (d, JP-C = 12.5), 131.04 (d, JP-C = 2.0), 130.96 (d, JP-C = 8.5), 130.56 (d, JP-C = 40.9), 129.32 (d, JP-C = 1.9), 128.51 (d, JP-C = 10.1), 127.78 (d, JP-C = 8.73), 125.86 (s), 117.47 (s), 104.90 (s), 73.66 (d, JP-C = 12.1), 71.51 (s), 67.08 (s), 59.75 (d, JP-C = 3.2), 31.30 (s), 29.69 (s), 19.06 (d, JP-C = 30.3 Hz), 11.62 (s), 2.89 (s) and 2.69 (s). 31P NMR (CDCl3): δ 31.4 (s) and −144.4 (sept, JP-F = 712 Hz). FAB MS: m/z 576 (M+ − PF6). Calc. for C29H34F6N2O2P2Ru: C, 48.61; H, 4.36; F, 15.91; N, 3.91; P, 8.64. Found: C, 48.46; H, 4.63; F, 15.69; N, 3.71; P, 8.56%.
/cm−1]: 1713 (CO). 1H NMR (CDCl3): δ 7.51–7.23 (m, 10 H), 5.10 (s, 1 H), 4.94 (s, 1 H), 4.95–4.87 (m, 1 H), 3.77–3.72 (m, 1 H), 2.96–2.93 (m, 1 H), 2.72–2.68 (m, 1 H), 2.19 (d, 3 H, JP-H = 1.5), 2.12 (s, 3 H), 2.10 (d, 3 H, JP-H = 1.2 Hz) and 1.71 (s, 3 H). 31P NMR (CDCl3): δ 43.53 (s) and −144.35 (sept, JP-F = 712 Hz). FAB MS: m/z 547 (M+ − PF6). Calc. for C27H30F6N2O2P2Ru: C, 46.89; H, 4.37; F, 16.48; N, 4.05; P, 8.96. Found: C, 46.69; H, 4.29; F, 16.55; N, 4.18; P, 9.02%.
/cm−1]: 1695 (CO). 1H NMR (acetone-d6): δ 7.61–7.57 (m, 4 H), 7.50–7.30 (m, 6 H), 7.10–7.04 (m, 3 H), 6.86 (t, 2 H, J = 8.1 Hz), 5.32 (s, 1 H), 5.13–5.03 (m, 1 H), 4.90 (s, 1 H), 4.10–4.02 (m, 1 H), 2.64 (s, 3 H), 2.47 (s, 3 H) and 2.28 (s, 3 H). 13C NMR (CDCl3): δ 212.56 (s), 166.98 (s), 135.51 (s), 134.61 (d, JP-C = 38.0), 134.09 (d, JP-C = 37.1), 133.26 (d, JP-C = 7.4), 130.64 (br), 128.76 (s), 128.67 (d, JP-C = 10.0), 127.79 (s), 127.27 (s), 125.40 (d, JP-C = 8.5), 106.40 (s), 95.13 (s), 76.84 (s), 76.32 (d, JP-C = 14.6), 72.29 (s), 59.10 (s), 38.30 (s), 19.79 (d, JP-C = 27.9 Hz) and 13.86 (s). 31P NMR (acetone-d6): δ 47.55 (s). FAB MS: m/z 633 (M+). Calc. for C30H30NO2PRuS2: C, 56.95; H, 4.78; N, 2.20; P, 4.89; S, 10.13. Found: C, 56.68; H, 4.64; N, 2.50; P, 4.73; S, 10.09%.
/cm−1]: 1732 (CO). 1H NMR (CDCl3): δ 8.63 (d, 1 H, J = 4.9), 7.82 (dd, 1 H, J = 7.9, 6.9), 7.45 (d, 1 H, J = 7.9), 7.33 (dd, 1 H, J = 4.9, 6.9), 6.08 (s, 6 H), 5.79 (s, 1 H), 5.66 (s, 1 H), 5.45 (d, 1 H, J = 12.9), 5.30 (d, 1 H, J = 12.9 Hz), 2.28 (s, 3 H) and 2.06 (s, 3 H). FAB MS: m/z 468 (M+ − PF6). Calc. for C20H20F6NO2PRu: C, 43.49; H, 3.65; F, 20.63; N, 2.54; P, 5.61. Found: C, 43.32; H, 3.57; F, 20.58; N, 2.55; P, 5.58%.
/cm−1]: 1737 (CO). 1H NMR (CDCl3): δ 8.68 (d, 1 H, J = 1.5), 7.89 (m, 1 H), 7.72 (m, 2 H), 7.58 (d, 1 H, J = 7.9), 7.41 (m, 4 H), 6.57 (s, 1 H), 6.40 (s, 1 H), 6.29 (s, 6 H), 5.52 (d, 1 H, J = 13.5), 5.40 (d, 1 H, J = 13.5 Hz) and 2.43 (s, 3 H). FAB MS: m/z 170 (M+ − PF6). Calc. for C25H22F6NO2PRu: C, 48.87; H, 3.65; F, 18.55; N, 2.28; P, 5.04. Found: C, 48.92; H, 3.41; F, 18.72; N, 2.21; P, 5.10%.
/cm−1]: 1714 (CO). 1H NMR (CD3CN): δ 8.53 (d, 1 H, J = 5.1), 7.66 (dd, 1 H, J = 7.5, 7.4), 7.41 (d, 1 H, J = 7.5), 7.27 (t, 1 H, J = 7.4, 5.1), 5.28 (d, 1 H, J = 1.3), 5.22 (d, 1 H, J = 1.3 Hz), 4.60 (s, 1 H), 4.10 (s, 1 H), 1.95 (s, 3 H) and 1.72 (s, 3 H). Calc. for C18H20F6N3O2PRu: C, 38.86; H, 3.62; F, 20.49; N, 7.55; P, 5.57. Found: C, 38.97; H, 3.64; F, 20.90; N, 7.98; P, 5.38%.
/cm−1]: 1715 (CO). 1H NMR (CD3CN): δ 8.55 (d, 1 H, J = 4.9 Hz), 7.79 (m, 1 H), 7.47 (m, 3 H), 7.33 (m, 4 H), 7.29 (m, 4 H), 5.46 (s, 1 H), 4.79 (s, 1 H), 3.61 (m, 2 H) and 2.06 (s, 3 H). An analytically pure sample was not obtained due to low thermal stability of 4b.
/cm−1]: 1694 (CO). 1H NMR (acetone-d6): δ 10.10 (d, 1 H, J = 5.4), 10.00 (d, 1 H, J = 6.1), 9.14 (d, 1 H, J = 5.1), 8.55 (d, 1 H, J = 7.8), 8.49 (d, 1 H, J = 8.2), 8.30 (dd, 1 H, J = 7.8, 6.8), 8.20 (dd, 1 H, J = 7.2, 6.8), 8.02 (dd, 1 H, J = 7.1, 6.7), 7.91 (dd, 1 H, J = 7.3, 6.7), 7.82 (dd, 1 H, J = 7.8, 6.6), 7.61 (d, 1 H, J = 8.1), 7.27 (dd, 1 H, J = 6.2, 6.3), 5.72 (d, 1 H, J = 14.5), 5.11 (s, 1 H), 4.84 (s, 1 H), 4.55 (d, 1 H, J = 14.5 Hz), 1.64 (s, 3 H) and 1.48 (s, 3 H). FAB MS: m/z 485 (M+ − PF6). Calc. for C24H22F6N3O2PRu: C, 45.72; H, 3.52; F, 18.08; N, 6.66; P, 4.91. Found: C, 45.56; H, 3.28; F, 18.00; N, 6.57; P, 4.90%.
/cm−1]: 1721 (CO). 1H NMR (CDCl3): δ 7.48–7.43 (m, 4 H), 7.38–7.26 (m, 6 H), 6.08 (s, 6 H), 5.64 (s, 1 H), 5.41 (s, 1 H), 4.47–4.40 (m, 2 H), 2.56–2.50 (m, 2 H), 2.29 (d, 3 H, J = 2.2) and 2.25 (d, 3 H, J = 2.2 Hz). 31P NMR (CDCl3): δ
−20.89 (s) and −142.67 (sept, J = 712 Hz). FAB MS: m/z 529 (M+ − PF6). Calc. for C28H28F6O2P2Ru: C, 49.93; H, 4.19; F, 16.92; P, 9.20. Found: C, 49.92; H, 4.08; F, 16.73; P, 9.36%.
/cm−1]: 1724 (CO). 1H NMR (CDCl3): δ 7.52–7.27 (m, 15 H), 6.26 (s, 1 H), 5.98 (s, 1 H), 5.97 (s, 6 H), 4.54–4.48 (m, 2 H), 2.66–2.50 (m, 2 H) and 2.37 (s, 3 H). 31P NMR (CDCl3): δ
−17.10 (s) and −138.99 (sept, JP-F = 708 Hz). FAB MS: m/z 591 (M+ − PF6). Calc. for C25H22F6NO2PRu: C, 48.87; H, 3.65; F, 18.55; N, 2.28; P, 5.04. Found: C, 48.92; H, 3.41; F, 18.72; N, 2.21; P, 5.10%.
/cm−1]: 1715 (CO). 1H NMR (acetone-d6): δ 7.60–7.49 (m, 4 H), 7.40 (d, 6 H, J = 8.3 Hz), 6.42 (s, 6 H), 5.75 (s, 1 H), 5.68 (s, 1 H), 4.49–4.43 (m, 2 H), 2.70–2.60 (m, 2 H), 2.30 (s, 3 H) and 1.20 (s, 9 H). 31P NMR (acetone-d6): δ
−17.27 (s) and −138.99 (sept, JP-F = 708 Hz). FAB MS: m/z 572 (M+ − PF6). Calc. for C31H34F6O2P2Ru: C, 52.03; H, 4.79; F, 15.93; P, 8.66. Found: C, 51.77; H, 4.75; F, 16.00; P, 8.77%.
/cm−1]: 1732 (CO). 1H NMR (CDCl3): δ 7.45–7.34 (m, 10 H), 6.03 (s, 6 H), 5.68 (s, 1 H), 5.65 (s, 1 H), 4.35–4.32 (m, 2 H), 2.25 (s, 3 H), 2.15–2.04 (m, 2 H), 2.04 (s, 3 H) and 1.85 (m, 2 H). 31P NMR (acetone-d6): δ
−11.78 (s) and −144.35 (sept, JP-F = 712 Hz). FAB MS: m/z 543 (M+ − PF6). Calc. for C29H30F6O2P2Ru: C, 50.66; H, 4.40; F, 16.58; P, 9.01. Found: C, 50.40; H, 4.21; F, 16.50; P, 9.22%.
/cm−1]: 1715 (CO). 1H NMR (CDCl3): δ 7.58–7.45 (m, 5 H), 7.31–7.25 (m, 3 H), 6.90 (t, 2 H, J = 8.1), 5.14 (s, 1 H), 5.06–4.97 (m, 1 H), 4.22 (s, 1 H), 3.97–3.90 (m, 1 H), 2.83–2.75 (m, 1 H), 2.57–2.50 (m, 1 H), 2.16 (d, 3 H, JP-H = 1.0), 2.11 (d, 3 H, JP-H = 1.2 Hz), 2.01 (s, 3 H) and 1.84 (s, 3 H). 13C NMR (acetone-d6): δ 166.52 (s), 135.06 (d, JP-C = 38.2), 133.95 (d, JP-C = 12.4), 131.20 (d, JP-C = 2.4), 130.41 (d, JP-C = 8.1), 130.08 (d, JP-C = 42.2), 129.20 (d, JP-C = 1.9), 128.96 (d, JP-C = 10.4), 127.85 (d, JP-C = 12.4), 126.89 (s), 125.61 (s), 108.17 (s), 98.41 (s), 77.79 (d, JP-C = 12.4), 74.47 (s), 68.81 (s), 59.21 (d, JP-C = 3.7), 17.72 (d, JP-C = 30.5 Hz), 11.70 (s), 11.22 (s), 2.82 (s) and 2.60 (s). 31P NMR (CDCl3): δ 35.56 (s) and −142.92 (sept, JP-F = 712 Hz). FAB MS: m/z 533 (M+ − PF6). Calc. for C28H28F6N2O2P2Ru: C, 46.09; H, 4.17; F, 16.32; N, 4.16; P, 9.14. Found: C, 45.87; H, 3.91; F, 16.66; N, 4.30; P, 9.00%.
/cm−1]: 1727 (CO). 1H NMR (CDCl3): δ 7.94–7.70 (m, 2 H), 7.56–7.48 (m, 3 H), 7.42–7.35 (m, 3 H), 7.06 (t, 2 H, J = 8.1), 5.18 (d, 1 H, J = 1.7), 5.05–4.96 (m, 1 H), 4.46 (d, 1 H, J = 1.7), 3.93–3.87 (m, 1 H), 2.89–2.81 (m, 1 H), 2.57–2.50 (m, 1 H), 2.18 (d, 3 H, JP-H = 1.0), 2.16 (d, 3 H, JP-H = 1.5 Hz), 2.06 (s, 3 H) and 1.26 (s, 9 H). 13C NMR (acetone-d6): δ 167.61 (s), 134.42 (d, JP-C = 38.9), 134.13 (d, JP-C = 12.5), 131.04 (d, JP-C = 2.0), 130.96 (d, JP-C = 8.5), 130.56 (d, JP-C = 40.9), 129.32 (d, JP-C = 1.9), 128.51 (d, JP-C = 10.1), 127.78 (d, JP-C = 8.73), 125.86 (s), 117.47 (s), 104.90 (s), 73.66 (d, JP-C = 12.1), 71.51 (s), 67.08 (s), 59.75 (d, JP-C = 3.2), 31.30 (s), 29.69 (s), 19.06 (d, JP-C = 30.3 Hz), 11.62 (s), 2.89 (s) and 2.69 (s). 31P NMR (CDCl3): δ 31.4 (s) and −144.4 (sept, JP-F = 712 Hz). FAB MS: m/z 576 (M+ − PF6). Calc. for C29H34F6N2O2P2Ru: C, 48.61; H, 4.36; F, 15.91; N, 3.91; P, 8.64. Found: C, 48.46; H, 4.63; F, 15.69; N, 3.71; P, 8.56%.
/cm−1]: 1713 (CO). 1H NMR (CDCl3): δ 7.51–7.23 (m, 10 H), 5.10 (s, 1 H), 4.94 (s, 1 H), 4.95–4.87 (m, 1 H), 3.77–3.72 (m, 1 H), 2.96–2.93 (m, 1 H), 2.72–2.68 (m, 1 H), 2.19 (d, 3 H, JP-H = 1.5), 2.12 (s, 3 H), 2.10 (d, 3 H, JP-H = 1.2 Hz) and 1.71 (s, 3 H). 31P NMR (CDCl3): δ 43.53 (s) and −144.35 (sept, JP-F = 712 Hz). FAB MS: m/z 547 (M+ − PF6). Calc. for C27H30F6N2O2P2Ru: C, 46.89; H, 4.37; F, 16.48; N, 4.05; P, 8.96. Found: C, 46.69; H, 4.29; F, 16.55; N, 4.18; P, 9.02%.
/cm−1]: 1695 (CO). 1H NMR (acetone-d6): δ 7.61–7.57 (m, 4 H), 7.50–7.30 (m, 6 H), 7.10–7.04 (m, 3 H), 6.86 (t, 2 H, J = 8.1 Hz), 5.32 (s, 1 H), 5.13–5.03 (m, 1 H), 4.90 (s, 1 H), 4.10–4.02 (m, 1 H), 2.64 (s, 3 H), 2.47 (s, 3 H) and 2.28 (s, 3 H). 13C NMR (CDCl3): δ 212.56 (s), 166.98 (s), 135.51 (s), 134.61 (d, JP-C = 38.0), 134.09 (d, JP-C = 37.1), 133.26 (d, JP-C = 7.4), 130.64 (br), 128.76 (s), 128.67 (d, JP-C = 10.0), 127.79 (s), 127.27 (s), 125.40 (d, JP-C = 8.5), 106.40 (s), 95.13 (s), 76.84 (s), 76.32 (d, JP-C = 14.6), 72.29 (s), 59.10 (s), 38.30 (s), 19.79 (d, JP-C = 27.9 Hz) and 13.86 (s). 31P NMR (acetone-d6): δ 47.55 (s). FAB MS: m/z 633 (M+). Calc. for C30H30NO2PRuS2: C, 56.95; H, 4.78; N, 2.20; P, 4.89; S, 10.13. Found: C, 56.68; H, 4.64; N, 2.50; P, 4.73; S, 10.09%.
X-Ray diffraction analysis for complex 10
Crystals suitable for X-ray diffraction were mounted on a glass fiber with epoxy resin. All measurements were performed on a Rigaku AFC5R automated four circle diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71069 Å) at 23 °C. Three standard reflections were monitored at every 150 and no decay was observed. An empirical absorption correction was made by the ψ-scan technique.The structure was solved by Patterson methods (DIRDIF 94 Patty).18 All non-hydrogen atoms were refined anisotropically by full-matrix least-squares minimizing Σw(|Fo| − |Fc|)2. The hydrogen atoms were included at the calculated positions (dC–H = 0.95 Å) and their parameters not refined. All calculations were performed using the TEXSAN crystallographic software package.19 Crystallographic data are listed in Table 2.
| Chemical formula | C30H30NO2PRuS2 |
| Formula weight | 632.74 |
| Crystal system | Triclinic |
| Space group | P 1 (no. 2) |
| a/Å | 12.346(3) |
| b/Å | 12.759(4) |
| c/Å | 9.569(2) |
| α/° | 105.55(2) |
| β/° | 91.92(2) |
| γ/° | 105.01(3) |
| V/Å3 | 1393.9(7) |
| Z | 2 |
| μ/cm−1 | 7.98 |
| Total data collected | 6678 |
| Unique data | 6393 (Rint = 0.017) |
| Observed data | 5452 (I > 3.0σ(I)) |
| R | 0.032 |
| R′ | 0.045 |
CCDC reference number 186/1734.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture. We are grateful to The Material Analysis Center, ISIR, Osaka University, for support of spectral and X-ray measurements as well as microanalyses.References
- S. G. Davies , Organotransition Metal Chemistry: Applications to Organic Synthesis, Pergamon, Oxford, 1982; Search PubMed; I. Ojima (Editor), Catalytic Asymmetric Synthesis, VCH, New York, 1993; Search PubMed; R. Noyori , Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994. Search PubMed.
- For reviews, see: H. Brunner, Adv. Organomet. Chem., 1980, 18, 151 Search PubMed; G. Consiglio and F. Morandini, Chem. Rev., 1987, 87, 761 CrossRef CAS; R. L. Halterman, Chem. Rev., 1992, 92, 965 CrossRef CAS; H. Brunner, Angew. Chem., Int. Ed., 1999, 38, 1195 CrossRef CAS.
- W. A. Schenk and M. Dürr, Chem. Eur. J., 1997, 3, 713 CrossRef CAS; W. A. Schenk, J. Frisch, M. Dürr, N. Burzlaff, D. Stalke, R. Fleischer, W. Adam, F. Prechtl and A. K. Smerz, Inorg. Chem., 1997, 36, 2372 CrossRef CAS; A. J. Davenport, D. L. Davies, J. Fawcett, S. A. Garratt, L. Lad and D. R. Russell, Chem. Commun., 1997, 2347 RSC; D. Carmona, F. J. Lahoz, S. Elipe, L. A. Oro, M. P. Lamata, F. Viguri, C. Mir, C. Cativiela and M. P. L.-R. de Víu, Organometallics, 1998, 17, 2986 CrossRef CAS; E. P. Kündig, C. M. Sauden and G. Bernardinelli, Angew. Chem., Int. Ed., 1999, 38, 1220 CAS.
- H. Brunner, K. Fisch, P. G. Jones and J. Salbeck, Angew. Chem., Int. Ed. Engl., 1989, 28, 1521 CrossRef; S. G. Davies, Aldrichchim. Acta, 1990, 23, 31 Search PubMed; M. Brookhart, Y. Liu, E. W. Goldmman, D. A. Timmers and G. D. Williams, J. Am. Chem. Soc., 1991, 113, 927 CrossRef CAS; J. W. Faller, M. R. Mazzieri, J. T. Nguyen, J. Parr and M. Tokunaga, Pure Appl. Chem., 1994, 66, 1463 CAS; J. A. Gladysz and B. J. Boone, Angew. Chem., Int. Ed. Engl., 1997, 36, 550 CrossRef.
- For reviews, see: H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS; A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587 CrossRef CAS.
- Y. Kataoka, Y. Saito, K. Nagata, K. Kitamura, A. Shibahara and K. Tani, Chem. Lett., 1995, 833 CAS; Y. Kataoka, Y. Saito, A. Shibahara and K. Tani, Chem. Lett., 1997, 621 CrossRef CAS; T. A. Mobley and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 3253 CrossRef CAS; Y. Kataoka, A. Shibahara, Y. Saito, T. Yamagata and K. Tani, Organometallics, 1998, 17, 4338 CrossRef CAS; B. M. Trost, B. Vidal and M. Thommen, Chem. Eur. J., 1999, 5, 1055 CrossRef CAS.
- J. C. Ruble and G. C. Fu, J. Org. Chem., 1996, 61, 7230 CrossRef CAS; P. I. Dosa, J. C. Ruble and G. C. Fu, J. Org. Chem., 1997, 62, 444 CrossRef CAS; J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492 CrossRef CAS; J. C. Ruble, J. Tweddell and G. C. Fu, J. Org. Chem., 1998, 63, 2794 CrossRef CAS; M. M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 10270 CrossRef CAS; J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532 CrossRef CAS; B. L. Hodous, J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1999, 121, 2637 CrossRef CAS.
- M. Uno, K. Ando, N. Komatsuzaki and S. Takahashi, J. Chem. Soc., Chem. Commun., 1992, 964 RSC; M. Uno, K. Ando, N. Komatsuzaki, T. Tanaka, M. Sawada and S. Takahashi, J. Chem. Soc., Chem. Commun., 1993, 1549 RSC; M. Uno, N. Komatsuzaki, K. Shirai and S. Takahashi, J. Organomet. Chem., 1993, 462, 343 CrossRef CAS; M. Uno, K. Ando, N. Komatsuzaki, T. Tsuda, T. Tanaka, M. Sawada and S. Takahashi, J. Organomet. Chem., 1994, 473, 303 CrossRef CAS; N. Komatsuzaki, M. Uno, K. Shirai, T. Tanaka, M. Sawada and S. Takahashi, J. Organomet. Chem., 1995, 498, 53 CrossRef CAS; M. Uno, K. Shirai, K. Ando, N. Komatsuzaki, T. Tanaka, M. Sawada and S. Takahashi, Chem. Lett., 1995, 7 CAS; Y. Morimoto, K. Ando, M. Uno and S. Takahashi, Chem. Commun., 1997, 1795 RSC; T. Katayama, Y. Morimoto, M. Yuge, M. Uno and S. Takahashi, Organometallics, 1999, 18, 3087 CrossRef CAS.
- N. Komatsuzaki, M. Uno, H. Kikuchi and S. Takahashi, Chem. Lett., 1996, 677.
- For reviews, see: P. Jutzi and U. Siemeling, J. Organomet. Chem., 1995, 500, 175 CrossRef CAS; P. Jutzi and T. Redeker, Eur. J. Inorg. Chem., 1998, 663 CrossRef CAS.
- B. Thierrien and T. R. Ward, Angew. Chem., Int. Ed., 1999, 38, 405 CrossRef CAS; B. Thierrien, A. König and T. R. Ward, Organometallics, 1999, 18, 1565 CrossRef.
- T. P. Gill and K. R. Mann, Organometallics, 1982, 1, 485 CrossRef CAS.
- T. Ohnishi, Y. Miyaki, H. Asano and H. Kurosawa, Chem. Lett., 1999, 809 CrossRef CAS; B. Thierrien, T. R. Ward, M. Pilkingston, C. Hoffmann, F. Gilardoni and J. Weber, Organometallics, 1998, 17, 330 CrossRef.
- D. Cantreux, J. Gamet, R. Jacquier and J. Verducci, Tetrahedron, 1984, 40, 3087 CrossRef.
- S. G. Davies, J. P. Macnally and A. J. Smallridge, Adv. Organomet. Chem., 1990, 30, 1 Search PubMed.
- Q. J. McCubbin, F. J. Stoddart, T. Welton, A. J. P. White and D. J. Williams, Inorg. Chem., 1998, 37, 3753 CrossRef CAS.
- A. W. Cordes and M. Draganjac, Acta Crystallogr., Sect. C, 1988, 44, 363 CrossRef.
- P. T. Beurskens , G. Admiraal , G. Beurskens , W. P. Bosman , S. Garcia-Granda , R. O. Gould , J. M. M. Smits and C. Smykalla , DIRDIF 92, Technical Report of the Crystallographic Laboratory, University of Nijmegan, 1994. Search PubMed.
- TEXSAN, Structure Analysis Package, Molecular Structure Corporation, Houston, TX, 1985 and 1992. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |