Synthesis and characterisation of thiolato Schiff base nickel(II) complexes. X-Ray structures of Ni(η5-C5H5)(PPh3)(SC6H4N![[double bond, length half m-dash]](https://www.rsc.org/images/entities/h2_char_e006.gif) CHC6H4Br-4′) and Ni(η5-C5H5)(PBu3)(SC6H4NCHC6H4CH3-4′)
CHC6H4Br-4′) and Ni(η5-C5H5)(PBu3)(SC6H4NCHC6H4CH3-4′)![[hair space]](https://www.rsc.org/images/entities/h2_char_200a.gif) †
†
Funzani A.
Nevondo
,
Andrew M.
Crouch
and
James
Darkwa
*
Department of Chemistry, University of the Western Cape, Private Bag X17, Bellville, 7535, South Africa. E-mail: jdarkwa@uwc.ac.za
First published on 13th January 2000
Abstract
A series of complexes of formula Ni(η5-C5H5)(PR3)(SC6H4N![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CHC6H4X-4′) (R = Ph, Bu; X = F, Cl, Br, CH3, OH, H) has been isolated from the reactions of Ni(η5-C5H5)(PPh3)Cl or the bromo derivative, the thiol Schiff base ligands, 4-HSC6H4NCHC6H4X-4′, and triethylamine. Preliminary thermal analysis data indicate that the PBu3 complexes could exhibit liquid crystalline behaviour. Electrochemistry of the PPh3 compounds gave irreversible redox couples, in contrast to the quasi-reversibility of the PBu3 compounds.
CHC6H4X-4′) (R = Ph, Bu; X = F, Cl, Br, CH3, OH, H) has been isolated from the reactions of Ni(η5-C5H5)(PPh3)Cl or the bromo derivative, the thiol Schiff base ligands, 4-HSC6H4NCHC6H4X-4′, and triethylamine. Preliminary thermal analysis data indicate that the PBu3 complexes could exhibit liquid crystalline behaviour. Electrochemistry of the PPh3 compounds gave irreversible redox couples, in contrast to the quasi-reversibility of the PBu3 compounds.
Introduction
Early investigations of Schiff bases that contain imine nitrogen and anionic sulfur ligands centred on their use as biological models.1 These were usually metal complexes of the ligands, formed in situ from the condensation of 2-mercaptoaniline and a carbonyl compound in the presence of a metal ion.2 The ligands bind in a bidentate fashion through the nitrogen and the sulfur atoms. Without this in situ approach, the condensation products of the 2-mercaptoaniline and the carbonyl compound result in an undesirable cyclization to form heterocyclic compounds;![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 3 though the cyclized products exist in equilibrium with a small amount of a non-cyclized thiol compound.4 Some of the recent work on Schiff base ligands has however concentrated on their potential use in materials science; specifically liquid crystals5 and non-linear optical materials.6 In such applications, it is anticipated that the presence of a metal in a complex that contains a Schiff base would modify the properties of the complex. In particular, Schiff base ligands that can bind two metal centers as bridging ligands and posses delocalised π-electrons could function as conducting materials.
3 though the cyclized products exist in equilibrium with a small amount of a non-cyclized thiol compound.4 Some of the recent work on Schiff base ligands has however concentrated on their potential use in materials science; specifically liquid crystals5 and non-linear optical materials.6 In such applications, it is anticipated that the presence of a metal in a complex that contains a Schiff base would modify the properties of the complex. In particular, Schiff base ligands that can bind two metal centers as bridging ligands and posses delocalised π-electrons could function as conducting materials.
Our interest in the synthesis and charaterization of nickel(II) thiolato complexes7 has been extended to thiolato Schiff base ligands. We expect, in this approach, to use these Schiff base ligand complexes as building blocks for preparing compounds that could be used as liquid crystals and conducting materials. Schiff base complexes that behave as metallomesogens are of two types. The first type are complexes where the Schiff base is bound to a metal.8 This includes a number of tetradentate nickel Schiff base compounds9 (Chart 1). The second type are complexes where the Schiff base ligand is attached to a ligand bound to a metal via atoms other than the imine unit. These latter compounds are substituted ferrocenes, with one of the cyclopentadienyl ligands carrying the Schiff base6,10 (Chart 1). Apart from nickel Schiff base metallomesogens, nickel thiolates11,12 and dithiolene13 compounds also show a range of liquid crystalline behaviour. The dinuclear complexes, Ni2(S2CR)4 (R = CnH2n + 1), exhibit monotropic lamellar mesophases when n = 4 or 8.12
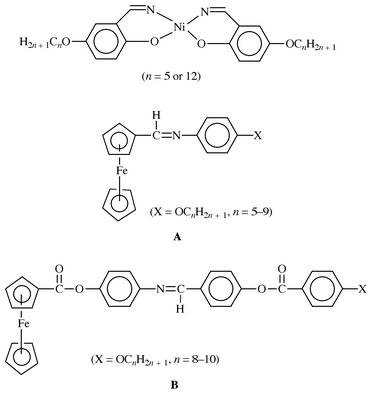 | ||
| Chart 1 | ||
In this project we have combined the potential of Schiff base ligands to form metallomesogens with that of the thiolate ligands to synthesize nickel thiolato Schiff base complexes. The result is the synthesis of a new class of nickel thiolato Schiff base compounds with potential mesogenic behaviour.
Experimental
Materials and instrumentation
All solvents were dried by procedures described as previously reported.7c All the commercially available chemicals were used as received. The nickel starting materials, Ni(η5-C5H5)(PPh3)Cl,14 Ni(η5-C5H5)(PPh3)Br14 and Ni(η5-C5H5)(PBu3)Cl,15 were prepared by the literature procedures. Infrared spectra were recorded on a Perkin-Elmer Paragon 1000PC FT spectrometer as Nujol mulls. The 1H, 13C and 31P NMR spectra were recorded on a Varian Gemini 2000 and referenced to residual CHCl3 for 1H (δ 7.26), 13C (δ 77.0) and to PPh3 (δ
−5.00) for 31P. Mass spectra of ligands were recorded on a Finnigan MAT GCQ GC/MS. The mass spectra of the metal complexes were recorded on a JEOL JMS-HX100 EBE in the FAB mode, using nitrobenzyl alcohol as the matrix as described previously.16 Thermal analyses were performed on a Perkin-Elmer TGA-7 and a Perkin-Elmer DSC-7 at a heating rate of 10 °C min−1 under nitrogen. Elemental analyses were performed in-house with a CARLO ERBA NA analyzer.
Electrochemical measurements
Cyclic voltammetric measurements were performed on a BAS-C50W electrochemical analyzer at room temperature, using sample concentrations of 10−3 M. Tetra-n-butylammonium tetrafluoroborate was used as supporting electrolyte. A three-electrode configuration, consisting of a platinum working electrode a platinum wire as auxiliary electrode and Ag/AgCl reference electrode was employed. The ferrocene/ferrocenium couple (+0.46 V vs. Ag/AgCl in CH2Cl2) was used as an internal standard. High purity and dry dichloromethane was used as the solvent and experiments were performed under dry nitrogen.Syntheses
CHC6H4X-4′ (X = F 1a, Cl 2a, Br 3a, CH34a, OH 5a, H 6a), were prepared by the following general procedure. To a solution of 4-aminothiophenol in ethanol (50 mL) was added the appropriate para-substituted benzaldehyde in a 1∶1 mole ratio and the solution stirred at room temperature for 6–24 h. The initial yellow solutions gave precipitates within 30 min, except for the hydroxy derivative, 5a. The precipitates were isolated by suction filtration and recrystallized from CH2Cl2–hexane. In isolating 5a, the clear solution was first evaporated to dryness and the residue recrystallized from CH2Cl2–hexane. The yields were generally moderate to high, 60–86%.
Complex 1a1a.. Yield: 63%. Anal. Calc. for C13H10FNS: C, 67.51; H, 4.36; N, 6.06. Found: C, 66.96; H, 4,41; N, 5.86%. 1H NMR (CDCl3): δ 8.41 (s, 1H, N
CH); 7.33 (m, 6H, SC6H4N CHC6H4F-4′); 6.59 (d, 2H, JHH = 8.80 Hz, SC6H4NCHC6H4F-4′); 3.49 (s, 1H, SH). IR (Nujol mull, cm−1): ν(CN) 1591. MS (EI): m/z = 231 (M+, 100%).
Complex 2a2a.. Yield: 71%. Anal. Calc. for C13H10ClNS: C, 63.03; H, 4.04; N, 5.65. Found: C, 63.19; H, 3.98; N, 5.63%. 1H NMR (CDCl3): δ 8.41 (s, 1H, N
CH); 7.42 (m, 6H, SC6H4NCHC6H4Cl-4′); 6.59 (d, JHH = 8.80 Hz, 2H, SC6H4N CHC6H4Cl-4′); 3.49 (s, 1H, SH). IR (Nujol mull, cm−1): ν(CN) 1591. MS (EI): m/z = 247 (M+, 100%).
Complex 3a3a.. Yield: 74%. Anal. Calc. for C13H10BrNS: C, 53.44; H, 3.42; N, 4.79. Found: C, 53.23; H, 3.49; N, 4.68%. 1H NMR (CDCl3): δ 8.39 (s, 1H, N
CH); 7.32 (m, 8H, SC6H4N CHC6H4Br-4′); 3.49 (s, 1H, SH). IR (Nujol mull, cm−1): ν(CN) 1590. MS (EI): m/z = 293 (M+, 100%).
Complex 4a4a.. Yield: 86%. Anal. Calc. for C14H13NS: C, 74.00; H, 5.72; N, 6.16. Found: C, 74.02; H, 6.12; N, 5.90%. 1H NMR (CDCl3): δ 8.39 (s, 1H, N
CH); 7.78 (d, JHH = 8.00 Hz, 2H, SC6H4NCHC6H4CH3-4′); 7.25 (m, 4H, SC6H4NCHC6H4CH3-4′); 6.58 (d, JHH = 8.80 Hz, 2H, SC6H4NCHC6H4CH3-4′); 3.47 (s, 1H, SH); 2.09 (s, 3H, SC6H4NCHC6H4CH3-4′). IR (Nujol mull, cm−1): ν(CN) 1602. MS (EI): m/z = 227 (M+, 100%).
Complex 5a5a.. Yield: 70%. Anal. Calc. for C13H11NOS: C, 68.12; H, 4.80; N, 6.11. Found: C, 66.91; H, 5.15; N, 6.14%. 1H NMR (d6-DMSO): δ 8.40 (s, 1H, N
CH); 7.50 (m, 8H, SC6H4NCHC6H4OH-4′); 3.50 (s, 1H, SH). IR (Nujol mull, cm−1): ν(CN) 1600. MS (EI): m/z = 229 (M+, 100%).
Complex 6a6a.. Yield: 75%. Anal. Calc. for C13H11NS: C, 73.23; H, 5.16; N, 6.57. Found: C, 73.13; H, 5.90; N, 6.57%. 1H NMR (CDCl3): δ 8.44 (s, 1H, N
CH); 7.88 (m, 2H, SC6H4N CHC6H5); 7.48 (m, 3H, SC6H4NCHC6H5); 7.32 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H5); 7.12 (d, JHH = 8.60 Hz, SC6H4N CHC6H5); 3.49 (s, 1H, SH). IR (Nujol mull, cm−1): ν(CN) 1597. MS (EI): m/z = 213 (M+, 100%).
Reaction of Ni(η5-C5H5)(PPh3)Br with 4-HSC6H4N![[double bond, length half m-dash]](https://www.rsc.org/images/entities/h3_char_e006.gif) CHC6H4X-4′: formation of Ni(η5-C5H5)(PPh3)(SC6H4NCHC6H4X-4′) (X = F 1b, Cl 2b, Br 3b, CH34b, OH 5b, H 6b)
CHC6H4X-4′: formation of Ni(η5-C5H5)(PPh3)(SC6H4NCHC6H4X-4′) (X = F 1b, Cl 2b, Br 3b, CH34b, OH 5b, H 6b)
In a typical reaction a mixture of Ni(η5-C5H5)(PPh3)Br (0.63 g, 1.29 mmol) and 1a ( 0.30 g, 1.28 mmol) was degassed. Addition of degassed toluene (75 mL), followed by Et3N (0.5 mL) immediately changed the solution from purple to brownish-green. After stirring for 4 h the mixture was filtered to remove solid Et3NHBr by-product. The filtrate was concentrated to about 20 mL; an equal volume of hexane was added and the solution cooled at −15 °C to precipitate the product. Recrystallization from CH2Cl2–hexane gave dark green crystalline 1b. Yield 62%. Anal. Calc. for C36H29FNPSNi: C, 70.15; H, 4.74; N, 2.27. Found: C, 70.08; H, 4.61; N, 2.35%. 1H NMR (CDCl3): δ 8.43 (s, 1H, NCH); 7.86 (dd, JHH = 8.40 Hz, JHF = 5.80 Hz, 2H, SC6H4NCHC6H4F-4′), 7.69 (m, 6H, PPh3); 7.39 (m, 11H, PPh3, SC6H4NCHC6H4F-4′); 7.13 (t, JHH/JHF = 8.20 Hz, 2H, SC6H4NCHC6H4F-4′); 6.87 (d, JHH = 8.40 Hz, 2H, SC6H4N CHC6H4F-4′); 5.14 (s, 5H, C5H5). 13C{1H} NMR (CDCl3): δ 154.1 (s, NCH); 144.2 (s); 132.2 (s); 131.7 (d, JCP = 42.0 Hz); 131.4 (s); 130.5 (s); 128.6 (s); 128.4 (s); 128.2 (d, JCP = 10.6 Hz); 126.4 (d, JCP = 41.0 Hz); 118.2 (s); 114.1 (s); 113.7 (s); 92.1 (s). 31P{1H} NMR (CDCl3): δ 35.38 (s, PPh3). IR (Nujol mull, cm−1): ν(CN) 1597. MS (FAB): m/z = 615 (M+, 8%).
All the PPh3 complexes were prepared and worked up in a similar manner as described above. Their analytical data were as reported below.
CH); 7.81 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4Cl-4′), 7.69 (m, 6H, PPh3); 7.38 (m, 13H, PPh3, SC6H4NCHC6H4Cl-4′); 6.87 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4Cl-4′); 5.14 (s, 5H, C5H5). 13C{1H} NMR (CDCl3): δ 156.6 (s, NCH); 146.6 (s); 145.2 (s); 137.4 (s); 135.9 (s); 134.8 (s); 134.4 (d, JCP = 42.0 Hz); 134.0 (s); 133.1 (s); 130.9 (d, JCP = 9.2 Hz); 129.7 (s); 129.0 (d, JCP = 41.0 Hz); 120.9 (s); 94.7 (s). 31P{1H} NMR (CDCl3): δ 35.37 (s, PPh3). IR (Nujol mull, cm−1): ν(CN) 1590. MS (FAB): m/z = 631 (M+, 3%).
CH); 7.71 (m, 8H, PPh3, SC6H4NCHC6H4Br-4′); 7.58 (d, JHH = 8.40 Hz, 2H, SC6H4N CHC6H4Br-4′); 7.38 (m, 11H, PPh3, SC6H4NCHC6H4Cl-4′); 6.87 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4Br-4′); 5.14 (s, 5H, C5H5). 13C{1H} NMR (CDCl3): δ 156.6 (s, NCH); 146.5 (s); 136.3 (s); 134.8 (s); 134.4 (d, JCP = 42.00 Hz); 134.0 (s); 133.1 (s); 132.7 (s); 130.9 (d, JCP = 9.00 Hz); 130.6 (s); 129.0 (d, JCP = 40.80 Hz); 125.9 (s); 120.9 (s); 94.7 (s). 31P{1H} NMR (CDCl3): δ 35.37 (s, PPh3). IR (Nujol mull, cm−1): ν(CN) 1587.
CH); 7.70 (m, 8H, PPh3, SC6H4NCHC6H4CH3-4′); 7.38 (m, 11H, PPh3, SC6H4NCHC6H4CH3-4′); 7.25 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4CH3-4′); 6.87 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4CH3-4′); 5.14 (s, 5H, C5H5); 2.39 (s, 3H, SC6H4NCHC6H4CH3-4′). 13C{1H} NMR (CDCl3): δ 157.7 (s, NCH); 143.4 (s); 134.2 (s); 133.7 (d, JCP = 42.40 Hz); 132.4 (s); 130.2 (d, JCP = 10.60 Hz); 129.5 (s); 128.5 (s); 128.3 (d, JCP = 40.80 Hz); 120.2 (s); 94.0 (s); 21.5 (s). 31P{1H} NMR (CDCl3): δ 35.41 (s, PPh3). IR (Nujol mull, cm−1): ν(CN) 1602. MS (FAB): m/z = 611 (M+, 15%).
CH); 7.69 (m, 8H, PPh3, SC6H4NCHC6H4OH-4′); 7.35 (m, 11H, PPh3, SC6H4NCHC6H4OH-4′); 7.25 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4OH-4′); 6.86 (dd, JHH = 8.60 Hz, JHH = 4.00 Hz, 4H, SC6H4N CHC6H4OH-4′); 5.13 (s, 5H, C5H5). 13C{1H} NMR (CDCl3): δ 154.2 (s, NCH); 136.1 (s); 136.0 (s); 132.3 (s); 131.7 (d, JCP = 42.40 Hz); 130.0 (s); 128.6 (s); 128.2 (s); 126.4 (d, JCP = 40.80 Hz); 118.1 (s); 113.8 (s); 92.1 (s). 31P{1H} NMR (CDCl3): δ 35.43 (s, PPh3). IR (Nujol mull, cm−1): ν(CN) 1604. MS (FAB): m/z = 613 (M+, 50%).
CH); 7.83 (m, 2H, SC6H4NCHC6H5); 7.69 (m, 6H, PPh3); 7.39 (m, 14H, PPh3, SC6H4NCHC6H5); 6.83 (d, JHH = 8.40 Hz, 2H, SC6H4N CHC6H5); 5.14 (s, 5H, C5H5). 13C{1H} NMR (CDCl3): δ 157.6 (s, NCH); 146.4 (s); 134.2 (s); 133.7 (d, JCP = 42.66 Hz); 133.4 (s); 132.5 (s); 132.2 (s); 130.2 (d, JCP = 10.60 Hz); 128.6 (d, JCP = 36.40 Hz); 128.3 (d, JCP = 40.00 Hz); 120 (s); 92.1 (s). 31P{1H} NMR (CDCl3): δ 35.36 (s, PPh3). IR (Nujol mull, cm−1): ν(CN) 1581.
Reaction of Ni(η5-C5H5)(PBu3)Cl with 4-HSC6H4NCHC6H4X-4′: formation of Ni(η5-C5H5)(PBu3)(SC6H4NCHC6H4X-4′) (X = F 1c, Cl 2c, Br 3c, CH34c, H 6c)
To a solution of 4-HSC6H4NCHC6H4F-4′
1a (0.35 g, 1.38 mmol) in toluene (25 mL) was added a solution of Ni(η5-C5H5)(PBu3)Cl (0.53 g, 1.38 mmol) in toluene (25 mL) via a pressure equalizing dropping funnel. Addition of excess Et3N (1.0 mL) gradually changed the purple solution to brown. After stirring the reaction mixture for 18 h, it was filtered to remove Et3NHCl as a by-product. The solvent was removed under reduced pressure and the residue recrystallized from CH2Cl2–hexane and isolated as dark brown crystalline 1c. Yield 69%. Anal. Calc. for C30H44FNPSNi: C, 64.79; H, 7.37; N, 2.52. Found: C, 64.02; H, 7.81; N, 2.57%. 1H NMR (CDCl3): δ 8.44 (s, 1H, NCH); 7.88 (dd, JHH = 8.80 Hz, JHF = 5.60 Hz, 2H, SC6H4NCHC6H4F-4′); 7.63 (d, JHH = 8.20 Hz, 2H, SC6H4N CHC6H4F-4′); 7.13 (t, JHF = 8.60, 2H, SC6H4NCHC6H4F-4′); 6.95 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4F-4′); 5.27 (s, 5H, C5H5); 1.43 (m, 18H, PBu3); 0.92 (t, JHH = 7.00 Hz, 9H, PBu3). 13C{1H} NMR (CDCl3): δ 155.9 (s, NCH), 145.9 (s); 144.7 (s); 133.7 (s); 133.0 (s); 130.4 (d, JCF = 86.60 Hz); 120.0 (s); 115.8 (d, JCF = 86.60 Hz); 91.8 (s); 26.3 (s); 24.5 (d, JCP = 112.00 Hz); 21.1 (d, JCP = 53.20 Hz); 13.7 (s). 31P{1H} NMR (CDCl3): δ 22.41 (s, PBu3). IR (Nujol mull, cm−1): ν(CN) 1601.
CH); 7.81 (d, JHH = 8.60 Hz, 2H, SC6H4NCHC6H4Cl-4′); 7.63 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4Cl-4′); 7.41 (d, JHH = 8.60 Hz, 2H, SC6H4N CHC6H4Cl-4′); 6.96 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4Cl-4′); 5.27 (s, 5H, C5H5); 1.48 (m, 18H, PBu3); 0.92 (t, JHH = 7.00 Hz, 9H, PBu3). 13C{1H} NMR (CDCl3): δ 155.7 (s, NCH); 145.2 (s); 136.7 (s); 135.3 (s); 133.7 (s); 129.0 (s); 128.9 (s); 120.3 (s); 91.9 (s); 26.3 (d, JCP = 4.60 Hz); 24.2 (d, JCP = 53.20 Hz); 22.5 (d, JCP = 110.60 Hz); 13.6 (s). 31P{1H} NMR (CDCl3): δ 22.43 (s, PBu3). IR (Nujol mull, cm−1): ν(CN) 1589.
CH); 7.81 (d, JHH = 8.60 Hz, 2H, SC6H4NCHC6H4Br-4′); 7.62 (d, JHH = 8.60 Hz, 2H, SC6H4NCHC6H4Br-4′); 7.42 (d, JHH = 8.40 Hz, 2H, SC6H4N CHC6H4Br-4′); 6.96 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4Br-4′); 5.27 (s, 5H, C5H5); 1.49 (m, 18H, PBu3); 0.93 (t, JHH = 7.00 Hz, 9H, PBu3). 13C{1H} NMR (CDCl3): δ 153.8 (s, NCH); 143.7 (s); 143.2 (s); 134.7 (s); 133.3 (s); 131.7 (s); 127.7 (s); 127.3 (s); 118.3 (s); 89.9 (s); 24.3 (s); 22.5 (d, JCP = 110.60 Hz); 22.2 (d, JCP = 53.20 Hz); 11.7 (s). 31P{1H} NMR (CDCl3): δ 22.45 (s, PBu3). IR (Nujol mull, cm−1): ν(CN) 1592.
CH); 7.76 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4CH3-4′); 7.61 (d, JHH = 8.60 Hz, 2H, SC6H4NCHC6H4CH3-4′); 7.25 (d, JHH = 8.00 Hz, 2H, SC6H4N CHC6H4CH3-4′); 6.95 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H4CH3-4′); 5.26 (s, 5H, C5H5); 2.40 (s, 3H, SC6H4NCHC6H4CH3-4′); 1.47 (m, 18H, PBu3); 0.92 (t, JHH = 7.00 Hz, 9H, PBu3). 13C{1H} NMR (CDCl3): δ 155.6 (s, NCH); 144.5 (s); 142.0 (s); 139.3 (s); 132.2 (s); 131.7 (s); 127.5(s); 126.6 (s); 118.3 (s); 89.9 (s); 24.3 (s); 24.3 (s); 22.5 (d, JCP = 28.22 Hz); 22.2 (d, JCP = 13.38 Hz); 19.5 (s); 11.7 (s). 31P{1H} NMR (CDCl3): δ 22.45 (s, PBu3). IR (Nujol mull, cm−1): ν(CN) 1607.
CH); 7.86 (d, JHH = 8.40 Hz, 2H, SC6H4NCHC6H5); 7.62 (d, JHH = 8.60 Hz, 2H, SC6H4N CHC6H5); 7.45 (t, JHH = 7.40 Hz, 3H, SC6H4NCHC6H5); 6.96 (d, JHH = 8.60 Hz, 2H, SC6H4NCHC6H5); 5.27 (s, 5H, C5H5); 1.54 (m, 18H, PBu3); 0.92 (t, JHH = 7.20 Hz, 9H, PBu3). 31P{1H} NMR (CDCl3): δ 22.41 (s, PBu3). IR (Nujol mull, cm−1): ν(CN) 1615.
Crystal structure determination of 3b
The structures of 3b and 4c were determined using a Siemens SMART and Enraf-Nonius Kappa diffractometers respectively, with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The crystal data, a summary of data collection and structure refinement are given in Table 1. The structures were solved by the Patterson method for primary atom sites and by difference map for atoms in secondary sites.17 All non-hydrogen atoms were refined anisotropically and hydrogen atoms were refined without restraints. The structures were refined by full-matrix least-squares on F![[hair space]](https://www.rsc.org/images/entities/i_char_200a.gif) 2 and calculations by either SHELXTL 9517 or SHELXL 97.18 In the refinement of 4c, the scale factor was allowed to vary by not setting a scale factor restraint. Absorption corrections were either fitted empirically by spherical harmonic functions19 or with the SORTAV20 programme.
2 and calculations by either SHELXTL 9517 or SHELXL 97.18 In the refinement of 4c, the scale factor was allowed to vary by not setting a scale factor restraint. Absorption corrections were either fitted empirically by spherical harmonic functions19 or with the SORTAV20 programme.
| 3b | 4c | |
|---|---|---|
| Formula | C36H29BrNPSNi | C31H47NPSNi |
| FW | 677.25 | 552.41 |
| T/K | 296(2) | 293(2) |
| λ/Å | 0.71073 | 0.71070 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/n | P 1 |
| a/Å | 10.833(5) | 10.809(1) |
| b/Å | 14.848(5) | 10.916(1) |
| c/Å | 19.742(9) | 14.430(1) |
| α/° | 80.813(2) | |
| β/° | 102.899(10) | 73.796(2) |
| γ/° | 68.446(2) | |
| V/Å3 | 3095.0(2) | 1517.5(1) |
| Z | 4 | 4 |
| D calc/Mg m−3 | 1.4531 | 1.209 |
| μ/mm−1 | 2.063 | 0.780 |
| F(000) | 1384 | 592 |
| Crystal size/mm | 0.80 × 0.54 × 0.06 | 0.21 × 0.35 × 0.28 |
| No. of reflections collected/unique | 17906/6929 | 13355/6780 |
| R(int.) | 0.045 | 0.044 |
| Data/restraints/parameters | 6929/0/486 | 8801/0/305 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0525, wR2 = 0.1363 | R 1 = 0.0651, wR2 = 0.1605 |
| R indices (all data) | R 1 = 0.0847, wR2 = 0.1568 | R 1 = 0.0794, wR2 = 0.1704 |
| Largest difference peak and hole/e Å−3 | 0.584, −0.462 | 3.0, −3.1 |
CCDC reference number 186/1731.
See http://www.rsc.org/suppdata/dt/a9/a906949f/ for crystallographic files in .cif format.
Results and discussion
Synthesis of complexes
The condensation of 4-aminothiophenol and different para-substituted benzaldehydes resulted in formation of thiol imines (L) (Scheme 1), capable of functioning as monodentate ligands via the sulfur atom.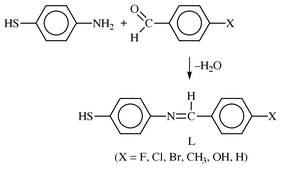 | ||
| Scheme 1 | ||
These thiol imines had characteristic νCN stretching frequencies in their infrared spectra. The nature of X, where X is a halide, affects the stretching frequency the most when the halide is fluorine. The more electronegative fluorine is expected to decrease the NCH double bond electron density, which should result in a lower νCN frequency. Hence it is not clear why the observed νCN for the fluoro compound is the highest. The chemical shifts observed in the 1H NMR spectra appear to be invariant for the compounds 1a–6a and show no effect of the substituents on the ligands. However, variation of X has some electronic influence on the electron density as shown by the electrochemical data (Table 2), discussed later.
The reactions of L with Ni(η5-C5H5)(PR3)Br (R = Ph, Bu), when the ligand was deprotonated by Et3N, at room temperature produced the thiolato complexes, Ni(η5-C5H5)(PR3)(SC6H4NCHC6H4X-4′) (Scheme 2). All the nickel complexes isolated, 1a–6b, were dark-brown to dark green crystalline solids. Spectroscopic characterization, elemental analyses, mass spectrometry and subsequent single crystal X-ray diffraction studies of Ni(η5-C5H5)(PPh3)(SC6H4NCHC6H4Br-4′) confirmed the products as formulated in Scheme 2.
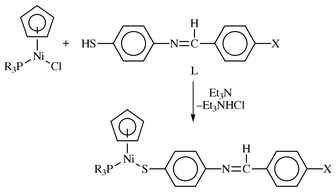 | ||
| Scheme 2 | ||
The 1H NMR spectra of complexes where X is a halide showed no significant chemical shift for the NCH proton in the free ligand and when complexed. A similar observation was made by Silver et al. for the iminyl protons of Fe(η5-C5H5)(η5-C5H4CHNC6H4Y-4′) and p-Me2NC6H4CHNC6H4Y-4′ (Y = F, Cl, Br21), where the iminyl protons have chemical shifts ranging from 8.38–8.43 ppm. This peak was also indifferent to the phosphine used. The observed peak values for both phosphine ligands were 8.43 ppm (PPh3) and 8.44 ppm (PBu3). The major differences in the 1H NMR spectra were found for the C5H5 ring chemical shifts. All the PPh3 compounds had essentially the same chemical shift of 5.14 ppm, whilst the PBu3 compounds had chemical shifts of 5.27 ppm. We reported a similar trend for 4-chlorothiophenolate complexes, Ni(η5-C5H5)(PR3)(SC6H4Cl-4′), with the same set of phosphines.7c This suggests that back-bonding from the Ni to PBu3 is more extensive than to PPh3. The back-bonding reduces the electron density on the nickel, which in turn draws electron density from the C5H5 ring and thus de-shields its protons. Further evidence of this is provided by the 31P NMR spectra, where the PBu3 peaks are more upfield than those of PPh3 despite the former being a better σ-donor. The chemical shift values of ca. 35.4 ppm and ca. 22.4 ppm for all the PPh3 and PBu3 complexes respectively point to the lack of any effect of the para-substituents of the thiolate ligand on the 31P NMR chemical shifts.
Each of the two phenyl groups in the thiolato ligands of the complexes exhibit a classical AA′BB′ spin system22 in the 1H NMR spectra and the spectrum of 2c depicts this clearly (Fig. 1). The 1H NMR spectral assignments for the C6-ring were made by reference to the para-fluoro derivative. This is facilitated by the hydrogen–fluorine coupling, affording an easy way to assign the ring protons based on the labeling in structure C. Protons Hd and Hd′ appear as a doublet of doublets, as a result of coupling of these protons with Hc and Hc′ and with the fluorine. Similarly, Hc and Hc′ are observed as a pseudo triplet from the same set of coupling. Thus for all complexes the most downfield doublets, doublet of doublets for the fluoro derivatives, are peaks of the Hd and Hd′ protons whilst Hc and Hc′ are the second most downfield peaks. By a similar argument the upfield doublets belong to Ha and Ha′.
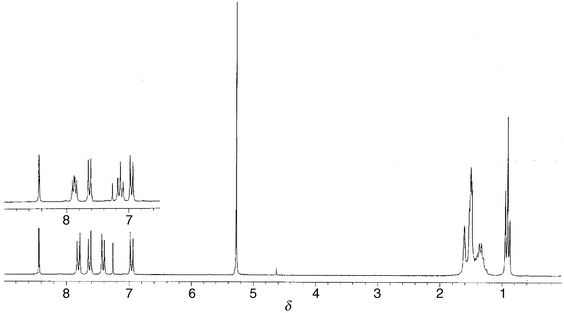 | ||
| Fig. 1 1H NMR spectra of 1c (insert) and 2c. | ||
When the mass spectra of compounds 1b–5b were run using the soft FAB ionisation technique, all the compounds except 3b showed molecular ions. However, even with the soft FAB ionization technique there was considerable fragmentation. All the compounds had fragments of m/z values of 385 and 647 as the dominant peaks in the spectra. These were identified as [(η5-C5H5)Ni(PPh3)]+ and [(η5-C5H5)Ni(PPh3)2]+ respectively. The latter ion is a product of a re-arrangement in the mass spectrometer. From thermal gravimetric analysis (TGA) (vide infra), it could be established that 3b readily decomposed at 180 °C whereas the rest of the complexes decompose above this temperature. It is therefore likely that the inability of 3b to give a molecular ion, even in the FAB mode, could be due to thermal decomposition.
Molecular structures of 3b and 4c
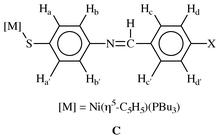
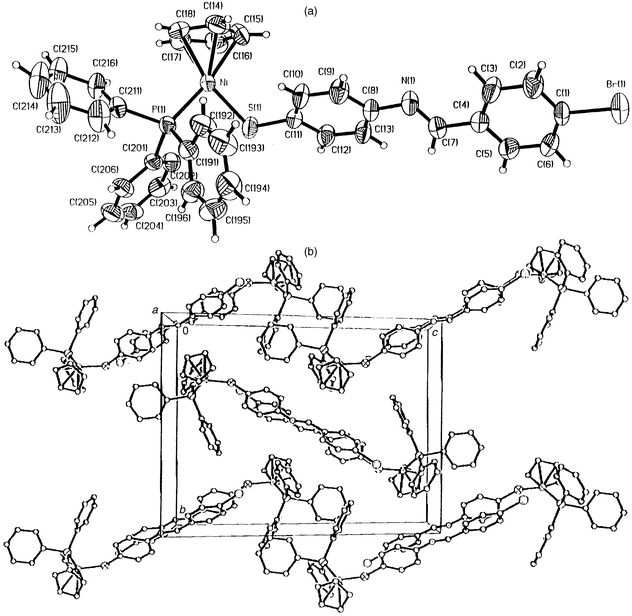
The proposed structures from the analytical data were confirmed by single crystal X-ray diffraction studies of 3b and 4c. The structures are depicted in Fig. 2 and 3, with selected bond distances and angles. Of particular note in the structure of 4c is a high residual electron density, about 0.3 Å from the nickel. This high residual electron density normally arises from inadequate treatment of data for absorption. But considering the absorption treatment applied to 4c, the high residual electron density may be due to the quality of the crystal. The structures are typical of the structural motifs found for Ni(η5-C5H5)(PR3)(EC6H4Cl-4) (R = Ph, Bu; E = S, Se; X = H, Cl).7a–c They include similarities in Cp–Ni, Ni–P and Ni–S bond distances. Comparison of the CN distances of the Schiff base ligands in 3b and 4c with the free thio Schiff base ligand, N,N′-bis(4-chlorobenzylidene)-2,2′-diaminodiphenyldisulfide23 similarly reveals no significant effect of the bonding interactions with the nickel. The CN bond distances in the free disulfide ligand are 1.261(5) and 1.258(5) Å respectively,23 whilst the CN distances in 3b and 4c are 1.261(5) and 1.262(7) Å respectively. The most striking features of the structures are found in the molecular packing [Fig. 2(b) and 3(b)]. Both have pair-wise association of molecules in a donor (D)–π-acceptor (A) DAAD type packing, with the cyclopentadienylnickel phosphino end of the molecule as donor and the Schiff base ligand as the acceptor. This DAAD type of packing is best depicted by 3b when the packing is viewed along the a-axis [Fig. 2(b)]. From this view the C6-ring arrangement of the thio ligand from each molecule are 4.5–5.2 Å apart.24 In both molecules the two C6-rings are in different planes, at right angles to each other. Another interesting feature of the packing is the orientation of the cyclopentadienyl rings. Again, in both molecules the cyclopentadienyl rings are oriented in different directions away from the pair-wise array of the rings. This allows the thio ligands to pack in an almost DAAD fashion in 3b, but in 4c the ring with the methyl substitution is bent away from the rest of the molecule to avoid steric crowding. The DAAD structural motif is similar to that observed in the ferrocenyl Schiff base compounds Fe(η5-C5H5)(η5-C5H4CHNC6H4NO2-4′).21 This type of stacking normally gives rise to C6-ring π–π interactions, which exists in the ferrocenyl compound above with ring distances of 3.478 Å. However, the inter-ring distances in 3b imply there is no such π–π interaction.
 | ||
| Fig. 2 (a) Crystal structure of 3b. Selected bond distances (Å) and angles (°) are: Ni–S(1) 2.1834(10), Ni–P 2.1447(10), Cp–Ni 1.743, C(11)–S(1) 1.766(4), C(7)–N(1) 1.261(5), C(1)–Br(1); P(1)–Ni–S(1) 91.18(4), Ni–S(1)–C(11) 112.04(13). (b) Packing diagram of 3b. | ||
 | ||
| Fig. 3 (a) Crystal structure of 4c. Selected bond distances (Å) and angles (°) are: Ni–S(1) 2.1483(12), Ni–P 2.1351(10), Cp–Ni 1.764, C(6)–S 1.752(5), C(12)–N 1.262(7), C(16)–C(19); P–Ni–S 92.32(5), Cp–Ni–P 132.8, Cp–Ni–S 134.8, Ni–S–C(6) 110.48(17). (b) Packing diagram of 4c. | ||
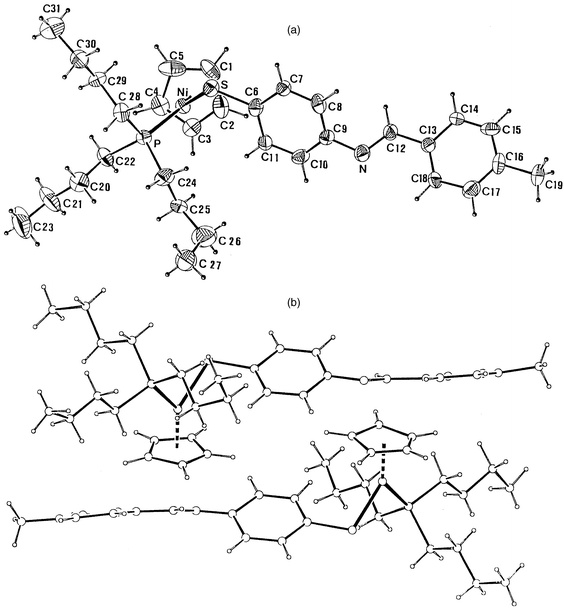
Electrochemical studies
Cyclic voltammograms of 1b–5b and 1c–4c were measured in CH2Cl2, with two representative voltammograms shown in Fig. 4. The results of the electrochemistry of the complexes are summarized in Table 2. Complexes 1b–5b showed irreversible redox behaviour, whereas 1c–4c were quasi-reversible. The triphenylphosphine complexes had ill-defined reduction peaks, whereas the tributylphosphine compounds exhibited well-defined reduction and oxidation peaks. The effect of the phosphine ligands on the electrochemical behaviour of the compounds was evident when triphenylphosphine was replaced with tributylphosphine (Fig. 4). Apart from 1c–4c being easier to oxidize, the quasi-reversibility of these compounds could be attributed to stabilization of the electroactive species by the more basic PBu3 as compared to PPh3. The voltammograms also indicate that a faster transfer kinetics is operational in complexes with the tributylphosphine ligand. The trend in the peak potentials of the halide derivatives (Table 2) was similar regardless of the phosphine. It appears the more electronegative halide derivatives were more difficult to oxidize, but it is difficult to see how these substituents could influence the ease of electron removal from the nickel centre. This trend, though, and the rest of the redox properties are similar to that found for Ni(η5-C5H5)(PR3)(SC6H4X-4′) (R = Ph, Bu; X = Cl, Br).7c | ||
| Fig. 4 Cyclic voltammograms of (a) 4b and (b) 4c. | ||
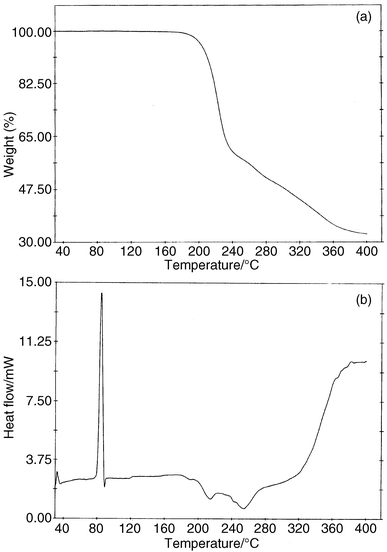
Thermal properties of 1c–4c and 3b
The thermal properties of selected complexes were investigated by thermogravimetric analysis (TGA) and by differential scanning calorimetry (DSC). Their thermal behaviour is typified by 4c (Fig. 5). Table 3 shows the rest of the thermal analysis data. All the complexes decomposed above 180 °C, except 3b, which decomposed at 180 °C. In the TGA, decomposition was generally via the loss of the thioimine ligand (L), except for 2c and 3b. The chloro derivative, 2c, showed loss of the cyclopentadienyl ligand; whereas 3b showed two successive losses of 4.80% and 20.78% that could not be attributed to any logical fragmentation sequence. The DSC data was obtained by two heating and cooling cycles to confirm the authenticity of the data. All complexes investigated gave endothermic peaks with ΔH values ranging from a low of 2.67 kJ mol−1 for 3b to a high of 46.22 kJ mol−1 for 1c (Table 3). The onset of the endothermic peaks in these complexes depends on two factors. First the PPh3 complex 3b had a higher onset temperature, accompanied by a low ΔH (Table 3). For the PBu3 complexes (1c–4c) the onset of the endothermic peak was at much lower temperatures and decreased with the electronegativity of the para-substituent. The lowest temperature was observed for the CH3 analogue (4c). This appears to indicate that the nickel end of the molecule needs electron density on the metal, whereas the other end is less electron rich. The high values of the endothermic peaks could be due to heat associated with mesophase changes.25 We are currently performing experiments to establish whether these complexes are indeed liquid crystalline.| Compounds | Weight lossa (%) |
Temperature rangeb/°C |
Peak valueb/°C |
ΔHb/kJ mol−1 |
|---|---|---|---|---|
| a Data from TGA (calculated weight loss in brackets). b Data from DSC. | ||||
| 3b | 4.80, 20.78 | 138.1–151.9 | 145.2 | 2.67 |
| 1c | 41.18 (43.58) | 102.2–114.9 | 109.6 | 46.22 |
| 2c | 11.78 (11.95) | 82.0–94.1 | 89.4 | 41.04 |
| 3c | 19.31, 16.71 | 78.5–93.6 | 88.4 | 28.16 |
| 4c | 43.49 (43.15) | 77.4–90.1 | 84.5 | 41.92 |
Conclusions
Schiff base nickel complexes that have the ligands bound to the metal via a sulfur atom are readily prepared. The PBu3 complexes show the onset of endothermic peaks in their DSC at temperatures ranging from 77.4 to 102.2 °C. Lower onset temperatures are associated with complexes containing less electron withdrawing groups in the 4′-position of the Schiff base ligand. The effect of the substituent can also be seen in the case of oxidation of the nickel centre as established from cyclic voltammetric studies. In this regard, the compounds with electron withdrawing substituents are more difficult to oxidise.Acknowledgements
Financial support from the National Research Foundation (NRF), South Africa, is gratefully acknowledged. One of us (F. A. N.) thanks the NRF and ESKOM, South Africa for bursary support. We are grateful to Ms. Leanne Cook, University of the Witwatersrand, South Africa and Dr John Bacsa, University of Cape Town, South Africa, who determined the structures as a service. Finally we wish to thank Ms. Ayesha Jacobs, University of Cape Town for the thermal analysis and Mr Tim Lesch, University of the Western Cape, for the elemental analysis.References
- (a) Y. Sugiura and Y. Hirayama, Inorg. Chem., 1976, 15, 679 CrossRef CAS; (b) L. Casella, Inorg. Chem., 1984, 23, 679 CrossRef.
- Y. Elerman and I. Svoboda, Acta Crystallogr., Sect. C, 1996, 52, 2705 CrossRef.
- (a) R. G. Charles and H. Freiser, J. Org. Chem., 1953, 18, 422 CrossRef CAS; (b) H. Jadamus, Q. Fernando and H. Freiser, J. Am. Chem. Soc., 1964, 86, 3056 CrossRef CAS; (c) Y. Sugiura, Y. Hirayama, T. Tanaka and K. Ishizu, J. Am. Chem. Soc., 1975, 97, 5577 CrossRef CAS; (d) S. Ide, S. G. Öztas, N. Ancin and M. Tüzün, Cryst. Res. Technol., 1996, 31, 427.
- L. F. Lindoy and S. E. Livingstone, Inorg. Chem., 1968, 7, 1149 CrossRef CAS.
- D. E. Wheeler, S. T. Hill, S. Milos, J. D. Williams and J. D. Johnson, J. Organomet. Chem., 1997, 530, 83 CrossRef CAS and references therein..
- W. E. Lindsell and L. Xinxin, J. Chem. Res. (S), 1998, 62 Search PubMed and references therein..
- (a) J. Darkwa, F. Bothata and L. M. Koczon, J. Organomet. Chem., 1993, 455, 235 CrossRef CAS; (b) J. Darkwa and W. Milius, Acta Crystallogr., Sect. C, 1996, 52, 2159 CrossRef; (c) J. Darkwa, R. M. Moutloali and T. Nyokong, J. Organomet. Chem., 1998, 564, 37 CrossRef CAS.
- (a) P. Espinet, M. A. Esteruelas, L. A. Oro, J. L. Serrano and E. Sola, Coord. Chem. Rev., 1992, 117, 215 CrossRef CAS; (b) S. A. Hudson and P. M. Maitlis, Chem. Rev., 1993, 93, 861 CrossRef CAS; (c) M. Ghedini, D. Pucci, A. Crispini and G. Baberio, Organometallics, 1999, 18, 2116 CrossRef CAS; (d) M. Lee, Y.-S. Yoo and M.-G. Choi, Macromolecules, 1999, 32, 2777 CrossRef CAS.
- R. Pache, H. Zaschke, A. Madicke. J. R. Chipperfield, A. B. Blake, P. G. Nelson and G. W. Gray, Mol. Liq. Cryst. Lett., 1988, 6, 81 Search PubMed.
- J. Malthête and J. Billard, Mol. Liq. Cryst. Lett., 1976, 34, 117 Search PubMed.
- K. Ohta, H. Ema, I. Yamamoto and K. Matsuzaki, Liq. Cryst., 1988, 3, 1671 CAS.
- K. Ohta, H. Ema, Y. Morizumi, T. Watanabe, T. Fujimoto and I. Yamamoto, Liq. Cryst., 1990, 3, 311.
- (a) A.-M. Giroud and U. T. Müller-Westerhoff, Mol. Cryst. Liq. Cryst., 1977, 41, 11 Search PubMed; (b) A.-M. Giroud, Ann. Phys. (Paris), 1978, 3, 47 Search PubMed; (c) A.-M. Giroud, A. Nazzal and U. T. Müller-Westerhoff, Mol. Cryst. Liq. Cryst., 1980, 56, 225 Search PubMed; (d) U. T. Müller-Westerhoff, A. Nazzal, R. J. Cox and A.-M. Giroud, Mol. Cryst. Liq. Cryst., 1980, 56, 249 Search PubMed; (e) M. Cortrait, J. Gaultier, C. Polycarpe, A.-M. Giroud and U. T. Müller-Westerhoff, Acta Crystallogr., Sect. C, 1983, 39, 833 CrossRef.
- K. W. Barnett, J. Chem. Educ., 1974, 51, 422 Search PubMed.
- J. Darkwa, Organometallics, 1994, 13, 4734.
- M. S. Thomas, J. Darkwa, E. Y. Osei-Twum and L. A. Latorja, Jr., Polyhedron, 1999, 18, 2803 CrossRef CAS.
- G. M. Sheldrick , SHELXTL 95, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1995. Search PubMed.
- G. M. Sheldrick , SHELXTL 97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997. Search PubMed.
- G. M. Sheldrick , SADABS, An Empirical Absorption Correction, Search PubMedprivate communication to subscribers of the Siemens CCD e-mail list..
- R. H. Blessing, J. Appl. Crystallogr., 1997, 30, 421 CrossRef CAS.
- A. Houlton, N. Jasim, R. M. G. Roberts, J. Silver, D. Cunningham, P. McArdle and T. Higgins, J. Chem. Soc., Dalton Trans., 1992, 2235 RSC.
- H. Freibolin , Basic one- and two-dimensional NMR spectroscopy, VCH, New York, ′nd edn., 1993, p. 121. Search PubMed.
- S. Ide, S. G. Öztas, N. Ancin and M. Tüzün, Acta Crystallogr., Sect. C, 1997, 53, 376 CrossRef.
- Inter-ring distances were determined with the program PLATON; A. L. Spek , University of Utrecht, The Netherlands, 1995. .
- M. E. Brown , Introduction to Thermal Analysis, Techniques and Applications, Chapman and Hall, London, 1988, p. 43. Search PubMed.
Footnote |
| † Supplementary data available: rotatable 3-D crystal structure diagram in CHIME format. See http://www.rsc.org/suppdata/dt/a9/a906949f/ |
| This journal is © The Royal Society of Chemistry 2000 |