Structural analysis and magnetic properties of the dicubane-like tetramer [Ni(dpk·OH)(N3)]4·2H2O (dpk = di-2-pyridyl ketone)†
Zurine E.
Serna
a,
M. Gotzone
Barandika
ab,
Roberto
Cortés
b,
M. Karmele
Urtiaga
c,
Gaston E.
Barberis
ad and
Teófilo
Rojo
*a
aDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad del País Vasco, Apdo. 644, Bilbao, 48080, Spain. E-mail: qiproapt@lg.ehu.es
bDepartamento de Química Inorgánica, Facultad de Farmacia, Universidad del País Vasco, Apdo. 450, Vitoria, 01080, Spain
cDepartamento de Mineralogía-Petrología, Facultad de Ciencias, Universidad del País Vasco, Apdo. 644, Bilbao, 48080, Spain
dInstituto de Física Gleb Wataghin, UNICAMP, 13087-970, Campinas (SP), Brazil
First published on 13th January 2000
Abstract
A nickel(II)tetramer with azide and di-2-pyridyl ketone (dpk) of general formula [Ni(dpk·OH)(N3)]4·2H2O (dpk·OH being the deprotonated gem-diol resulting from hydrolysis of dpk) was synthesized and structurally characterised through X-ray single crystal diffraction analysis and IR spectroscopy. The structure consists of dicubane-like tetrameric entities where simultaneous (1,1)-N3 and O bridges can be found, being an unprecedented arrangement for metal(II) azide systems. Measurements of the magnetic susceptibility revealed the occurrence of ferromagnetic interactions in the clusters. The extension of the magnetic exchange has been evaluated by means of a spin Hamiltonian with four J constants (J1 = 18.8, J2 = 6.9, J3 = 1.3 and J4 = 0.2 cm−1).
Introduction
Over the last years considerable research has been directed at the preparation of molecule-based magnets. Activity has been focused on obtaining nanoscale magnets in which each microcrystal behaves as a single domain. Nanoscale magnetic materials can be prepared by breaking down crystallites of bulk ferro- or ferri-magnetic samples to a much smaller size. However, this fragmentation gives rise to an undesirable distribution of particle sizes.1 A different way to deal with the preparation of nanomagnets, first suggested by Mataga,2 concerns single molecules having ground electronic states with a large number of unpaired electrons. On these bases, a great number of works dealing with transition metal clusters have been devoted to increasing the nuclearity of the systems.3An alternative approach to the generation of single-molecule type systems consists of enhancing the cluster anisotropy. Thus, due to its large single-ion zero-field splitting, NiII has been used to this purpose in some works.4 On the other hand, the nature and extension of the magnetic coupling are some of the features to be considered for the preparation of these systems. Thus, many of the clusters for nanomagnets involve intermetallic bridges through O atoms which, in most of the cases, provide modest values of the exchange coupling. In this sense, the use of ligands like azide can be expected to improve the coupling between metallic centres.
The nickel(II)-azide clusters reported so far,5 which are remarkably interesting from both the structural and the magnetic point of view, exhibit higher values of the J exchange constant than those found for O-bridged clusters. However, the latter have been much more extensively explored than the former. The reason for the scarce interest in nickel(II) azide systems for nanomagnets could be found in the poor nuclearity obtained up to now.
The above mentioned aspects suggest the need to explore the use of azide in this context. With this aim, this work has been focused on the NiII–azide–dpk system (dpk = di-2-pyridyl ketone). The dpk ligand has been observed easily to accommodate to steric requirements by co-ordinating in multiple fashions. This ligand (Scheme 1) has three potential donor sites, being able to chelate in bidentate (N,N and N,O) and tridentate (N,O,N) modes. Moreover, dpk has been observed occasionally to undergo hydration of the ketocarbonyl group forming a gem-diol which can co-ordinate either protonated (dpk·H2O) or deprotonated (dpk·OH).6
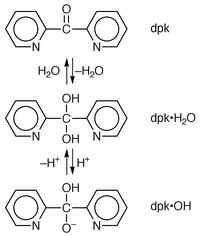 | ||
| Scheme 1 | ||
Our first results concern the preparation of a ferromagnetic cluster whose structure is unprecedented for metal(II) azide systems. Thus, this work reports on the magnetostructural characterisation of the tetrameric compound [Ni(dpk·OH)(N3)]4· 2H2O by means of X-ray diffraction analysis, IR spectroscopy and magnetic susceptibility measurements. Additionally, a theoretical interpretation of the magnetic behaviour of this compound is presented.
Experimental
Synthesis
The compound [Ni(dpk·OH)(N3)]4·2H2O was prepared in a diffusive cell with three compartments. The wing compartments contained an aqueous solution of Ni(NO3)2·6H2O (0.146 g, 0.5 mmol) and NaN3 (0.065 g, 1.0 mmol), and a methanolic solution of dpk (0.092 g, 0.5 mmol), respectively, while the central one contained methanol–water (1∶1). After several days, prismatic, green, X-ray quality single crystals (43% yield) were obtained. Elemental analysis and atomic absorption results were in good agreement with the C22H20N10Ni2O5 stoichiometry. Found (calc.%): 42.8 (42.58); H, 3.3 (3.25); N, 22.1 (22.58); Ni, 17.9 (48.69).CAUTION: azide salts are potentially explosive and should be handled in small quantities.
Physical measurements
Microanalyses were performed with a Perkin-Elmer 2400 analyser. Analytical measurements were carried out in an ARL 3410 + ICP with Minitorch equipment. IR spectroscopy was performed on a Nicolet 520 FTIR spectrophotometer in the 400–4000 cm−1 region. Magnetic susceptibilities of powdered samples were measured in the temperature range 1.8–300 K using a Quantum Design Squid magnetometer, equipped with a helium continuous-flow cryostat. The experimental susceptibilities were corrected for the diamagnetism of the constituent atoms (Pascal tables).Crystal structure determination
X-Ray measurements were made at room temperature on an Enraf-Nonius CAD-4 diffractometer with graphite-monochromated Mo-Kα radiation operating in ω–2θ scanning mode using suitable crystals for data collection. Accurate lattice parameters were determined from least-squares refinement of 25 well centred reflections. Intensity data were collected in the θ range 1–30°. During data collection, two standard reflections periodically observed showed no significant variation. Corrections for Lorentz-polarisation factors were applied to the intensity values. Owing to the large number of faces shown by the crystals no ideal method could have been used for absorption corrections and the program DIFABS![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 7 was applied.
7 was applied.
The structure was solved by heavy-atom Patterson methods using the program SHELXS 978 and refined by a full-matrix least-squares procedure on F2 using SHELXL 97.9 Non-hydrogen atomic scattering factors were taken from ref. 10. Crystallographic data and processing parameters are shown in Table 1. It is worth mentioning that the structure shows remarkable disorder affecting the azide ligands. Thus, the position of N9 and N10 atoms (corresponding to the terminal azides) has been split into two, A and B (with multiplicities of 0.5), for a better structural resolution.
| Formula | C22H20N10Ni2O5 | U/Å3 | 1357(1) |
| M | 621.90 | Z | 2 |
| Crystal system | Triclinic | T/°C | 25 |
| Space group |
P1 (no. 2) |
λ/Å | 0.71070 |
| a/Å | 10.230(3) | ρ obs/g cm−3 | 1.54(9) |
| b/Å | 10.358(4) | ρ calc/g cm−3 | 1.522 |
| c/Å | 10.396(8) | μ/mm−1 | 1.439 |
| α/° | 91.57(4) | Unique data | 7830 |
| β/° | 105.53(5) | Observed data | 7830 |
| γ/° | 96.26(3) |
R(R′)b |
0.0749(0.1951) |
CCDC reference number 186/1709.
See http://www.rsc.org/suppdata/dt/a9/a905430h/ for crystallographic files in .cif format.
Results and discussion
Structural analysis
The structure of [Ni(dpk·OH)(N3)]4·2H2O (Fig. 1) consists of centrosymmetric tetramers in which the nickel(II) ions are connected through μ-(1,1)-N3 and μ-O bridges. Additionally, there are two crystallisation molecules of water per tetrameric unit. Each cluster exhibits two end-on azides (co-ordinated by N5 and N5i atoms, respectively), two terminal azides (linked through N8 and N8i atoms, respectively) and four dpk·OH ligands (chelated by N1,O3,N2; N1i,O3i,N2i; N3,O1,N4 and N3i,O1i,N4i, respectively). Obviously, dpk·OH ligands are the result of the hydration and ulterior deprotonation of the original dpk groups (Scheme 1).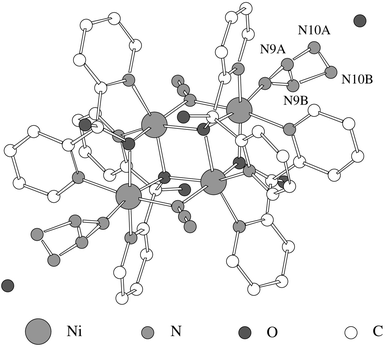 | ||
| Fig. 1 View of the tetrameric unit. | ||
The tetramers exhibit a dicubane-like core with two missing vertexes (Fig. 2) in which two types of octahedrally co-ordinated Ni atoms, Ni1 and Ni2, can be distinguished. Thus, the crystallographically related Ni2 and Ni2i occupy two vertexes of the common face of the dicubane unit, both metallic atoms being doubly O-bridged through O3 and O3i atoms (sited on the other two vertexes). These O atoms act as triple bridges since they are also bonded to N1 and N1i atoms, respectively, along the edges of both cubic subunits. Atom Ni1 is also doubly bridged to Ni2 (through O1) and to Ni2i (through N5end-on azide). Obviously, Ni1i is symmetrically bonded to Ni2 and Ni2i.
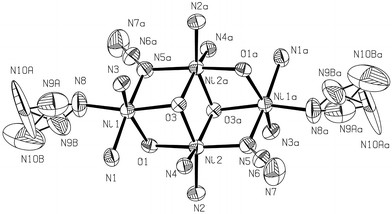 | ||
| Fig. 2 An ORTEP11 view (50% probability) of the dicubane-like core structure showing the co-ordination polyhedra in the tetrameric units.
| ||
The co-ordination spheres around both types of Ni are completed as follows. Considering that O1 and O3 atoms are located on the equatorial plane for Ni1, the remaining two positions are occupied by N3dpk·OH and N8terminal azide. On the other hand, besides the N5-bonded end on azide, located at one of the axial positions, N1dpk·OH can be found completing the octahedral sphere of Ni1. Describing now the co-ordination sphere around Ni2 also having O1 and O3 atoms on the equatorial plane, N5iend-on azide can be found occupying one of these positions, the fourth of them corresponding to N2dpk·OH. In the axial positions, besides the O3i atom, N4dpk·OH can be found. In this way, the intermetallic interaction through the azide and O bridges is reinforced by the N atoms of the N,O,N′-tridentate organic ligands.
The average distance between Ni and μ-O is 2.08(3) Å, while the Ni–Oμ3 average distance is 2.08(6) Å (Table 2). The Ni–O–Ni angles range from 94.84 to 99.68°. On the other hand, the Ni–Nazide average distance is 2.06(2) Å while the Ni–Nazide–Ni angle is 103.3(2). These latter values lie in the common range, 101–104°, for end-on azide bridged nickel(II) compounds. The average Ni⋯Ni distance through oxo-bridges is 3.10(4) Å, the distances through azide bridges being slightly longer (3.238(2) Å).
| Symmetry codes: (i) −x, −y, −z. | |||
|---|---|---|---|
| Ni1–N1 | 2.095(5) | Ni2–O1 | 2.105(4) |
| Ni1–N3 | 2.074(5) | Ni2–O3 | 2.030(4) |
| Ni1–N5 | 2.074(5) | Ni2–O3i | 2.107(4) |
| Ni1–O1 | 2.058(4) | N5–N6 | 1.192(7) |
| Ni1–O3 | 2.133(4) | N6–N7 | 1.120(8) |
| Ni1–N8 | 2.051(6) | N8–N9A | 1.11(4) |
| Ni2–N2 | 2.030(5) | N8–N9B | 1.09(3) |
| Ni2–N4 | 2.111(5) | N9A–N10A | 1.22(4) |
| Ni2–N5i | 2.058(5) | ||
| Ni1–O1–Ni2 | 94.8(2) | Ni2i–O3–Ni1 | 99.6(2) |
| Ni2–O3–Ni1 | 94.8(2) | N7–N6–N5 | 179.3(9) |
| Ni2–O3–Ni2i | 99.0(2) | N8–N9A–N10A | 170(4) |
| Ni2i–N5–Ni1 | 103.3(2) | ||
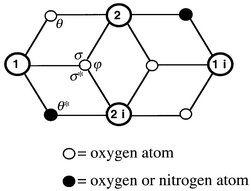
It is worth mentioning that similar dicubane-like cores have been found for other transition metal systems.4a,12Table 3 summarises some selected parameters for comparison between dicubane-like tetramers. Except from the present compound (which also exhibits Ni–Nend-on azide–Ni bridges), the rest of the compounds just show M–O–M interactions. All the tetramers in Table 3 are centrosymmetric and exhibit similar values of both the M⋯M distances and the M–O–M angles.
# which take place through N atoms
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tetramer | d(1–2) | d(1–2i) | d(2–2i) | d(1–li) | θ | θ* | σ | σ* | ϕ | Ref. |
| a [Ni4(H2O)2(PW9O34)2]10−. b [Mn(MeOH)L(OH)M(bpy)]4 (M = Ni, Mn or Cu; H4L = 1,2-bis(2-hydroxybenzamido)benzene); MnII and MII are located on sites 1 and 2, respectively. c [Cu4(tde)2(hfacac)4] (H2tde = 2,2′-thiodiethanol; Hhfacac = 1,1,1,5,5,5-hexafluoroacetylacetonate). d [Fe4(MeO)6(acac)4(N3)2] (acac = acetylacetonate). | ||||||||||
| Ni4 | 3.063 | 3.238# |
3.145 | 5.463 | 94.8 | 103.35# |
94.84 | 99.68 | 98.98 | This work |
| Ni4a |
3.124 | 3.109 | 3.196 | 5.352 | 97.6 | 99.0 | 92.7 | 93.4 | 95.5 | 4(a) |
| Ni2Mn2b |
3.203 | 3.195 | 3.112 | 5.590 | 102.7 | 102.0 | 94.2 | 92.0 | 98.1 | 12(b) |
| Mn4b |
3.211 | 3.112 | 2.744 | 5.846 | 99.8 | 99.7 | 97.8 | 99.3 | 93.1 | 12(b) |
| Cu2Mn2b |
3.367 | 2.744 | 2.954 | 6.048 | 97.3 | 97.6 | 99.9 | 100.1 | 96.9 | 12(b) |
| Cu4c |
3.121 | 2.954 | 3.230 | 5.458 | 108.8 | — | 90.6 | 111.4 | 93.8 | 12(a) |
| Fe4d |
3.212 | 3.230 | 3.214 | 3.214 | 108.4 | 108.0 | 96.8 | 96.7 | 100.6 | 12(c) |
IR Spectroscopy
A summary of the most important IR bands corresponding to the present compound together with their tentative assignment13 is described as follows. The compound shows an intense single band at 2071 cm−1 which is associated with the asymmetric stretching mode of the azide ligand. The splitting of this band is in accordance with the fact that two co-ordination fashions are present in this case. The absorption at about 1328 cm−1 is indicative of the end-on co-ordination of azide as this symmetric vibration mode is not usually active in end-to-end azides.In relation to the absorptions caused by the organic ligand, it is worth mentioning that the band at 1680 cm−1 attributed to the C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O bond in conjugation with the pyridyl rings in dpk is shifted to lower frequencies (1610 cm−1) as corresponds to a single C–O bond present after hydrolysis.14 Additionally, the IR spectrum revealed the bands corresponding to the skeleton vibrations of the co-ordinated dpk·OH which appear at slightly shifted frequencies in relation to free dpk. Thus, bands for co-ordinated dpk·OH (free dpk) are: 1540(1578/1545) cm−1 attributed to the pyridyl ring stretching; 1020(998) cm−1 for the pyridyl ring breathing; 757(753/742) cm−1 attributed to the pyridyl ring C–H out-of-plane bending and 660(662) cm−1 for the pyridyl ring in-plane vibration.
O bond in conjugation with the pyridyl rings in dpk is shifted to lower frequencies (1610 cm−1) as corresponds to a single C–O bond present after hydrolysis.14 Additionally, the IR spectrum revealed the bands corresponding to the skeleton vibrations of the co-ordinated dpk·OH which appear at slightly shifted frequencies in relation to free dpk. Thus, bands for co-ordinated dpk·OH (free dpk) are: 1540(1578/1545) cm−1 attributed to the pyridyl ring stretching; 1020(998) cm−1 for the pyridyl ring breathing; 757(753/742) cm−1 attributed to the pyridyl ring C–H out-of-plane bending and 660(662) cm−1 for the pyridyl ring in-plane vibration.
Magnetic properties
The magnetic characterisation was carried out through measurements of the thermal variation of the magnetic susceptibility, χm. The experimental χm values continuously increase upon cooling, becoming exponential for temperatures near 0 K.The experimental data plotted as the thermal variation of the reciprocal susceptibility, χm−1, and the product χmT are shown in Fig. 3. The variation of χm−1 is well described by the Curie–Weiss law down to 100 K. At lower temperatures χm−1 shows a characteristic curvature. The values Cm = 1.14 cm3 K mol−1 and g = 2.139 found are typical for octahedrally co-ordinated NiII.15 The Weiss temperature has been calculated to be θ = +30.8 K. The χmT magnitude continuously increases with decreasing temperature from 1.3 cm3 K mol−1 (per Ni atom) at RT to a maximum value of 2.62 cm3 K mol−1 at 12 K. After further cooling, the curve rapidly decreases tending to χmT = 0. These results are indicative of the occurrence of ferromagnetic coupling between metallic centres whose extension was theoretically estimated as described below.
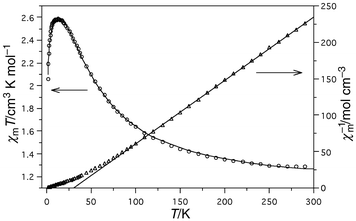 | ||
| Fig. 3 Thermal evolution of χm-1 and χmT and their corresponding theoretical curves. | ||
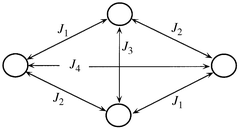
The theoretical approach to the magnetic behaviour of the compound has been carried out by considering the following Heisenberg Hamiltonian Ĥ = −2Σ4i,j = 1Jij![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) i·j. According to the ideal C2h symmetry, the exchange Hamiltonian, corresponding to four nickel(II) centres with S = 1 and a total degeneracy of (2S + 1)4 = 81, can be expressed as in eqn. (1) where the last term corresponds to the Zeeman interaction, J1 to the Ni–(Nend-on azide,O)–Ni bridge, J2 and J3 to Ni–(O,O)–Ni interactions and J4 to the exchange through the longest pathway.
i·j. According to the ideal C2h symmetry, the exchange Hamiltonian, corresponding to four nickel(II) centres with S = 1 and a total degeneracy of (2S + 1)4 = 81, can be expressed as in eqn. (1) where the last term corresponds to the Zeeman interaction, J1 to the Ni–(Nend-on azide,O)–Ni bridge, J2 and J3 to Ni–(O,O)–Ni interactions and J4 to the exchange through the longest pathway.
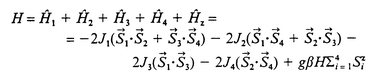
| (1) |
At this point, some considerations about the method employed for calculation of the thermally accessible spin levels should be made. Thus, these theoretical calculations are usually performed by means of Kambé’s vector coupling method16 as long as the exchange Hamiltonian is fully isotropic (Heisenberg model). In this way the Hamiltonian can be expressed through an appropriate set of intermediate spin operators that directly gives a diagonal eigenmatrix. Unfortunately, this method is restricted to high-symmetric systems which is not the current case. The procedure followed for calculation of the eigenvalues of the Hamiltonian for our compound is described below.
According to previous evidence in similar systems (Table 4), it can be concluded that the exchange interaction in eqn. (1) is the most important part of the magnetic Hamiltonian. Thus, approximate J values were calculated following well known numerical procedures,17 the order of magnitude of the exchange coupling parameters being J1 > J2 > J3 > J4 ≫ gβHM.
| Tetramer | Ni4 | Ni4 | Ni2Mn2 | Mn4 | Cu2Mn2 | Cu4 | Fe4 |
|---|---|---|---|---|---|---|---|
| a Corresponds to Ni–(N,O)–Ni pathways. The rest of them are M–(O,O)–M. b No fitting has been done but antiferromagnetic coupling has been observed. | |||||||
| Ref. | This work | 4(a) | 12(b) | 12(b) | 12(b) | 12(a) | 12(c) |
| J 1 | +18.8a |
J 1 = J2 = +6.5 | J 1 = J2 = −1.5 | J 1 = J2 = −3.5 | J 1 = J2 = −4.5 | b | b |
| J 2 | +6.9 | J 1 = J2 = +6.5 | J 1 = J2−1.5 | J 1 = J2 = −3.5 | J 1 = J2 = −4.5 | b | b |
| J 3 | +1.3 | 2.5 | −2.6 | −14.1 | −8.9 | b | b |
| J 4 | +0.2 | — | — | — | — | b | b |
With the aim of diagonalising the first term (Ĥ1) in eqn. (1), a basis for the coupled tetramer was selected. This provides 81 levels, degenerated in the projection of total moment as |ψi〉 = |
1,2,12,
3,4,34,,M〉. Thus, the individual spins are coupled in such a way that Ĥ1 is diagonal in this basis, while Ĥ2, Ĥ3 and Ĥ4 are non-diagonal. It should also be mentioned that the terms in those Hamiltonians do not mix subspaces with different values of the total S which can adopt the following values: S = 0, 1, 2, 3, 4 (with 3, 6, 6, 3 and 1 multiplicities, respectively). Additionally, the Zeeman interaction is diagonal in every basis since it was considered to be isotropic.
In order to solve the whole Hamiltonian, the selected basis was projected into bases that diagonalise each of the terms in eqn. (1). This was carried out by calculating the Clebsch–Gordan coefficients that change the basis and, afterwards, the whole matrix in the basis of eqn. (1). The Clebsch–Gordan coefficients were obtained by using 9 − j Wigner coefficients.18 The fact that the dimension for the S = 1 and S = 2 subspaces is six was the determining factor for the eigenmatrix to be numerically diagonalised. This was carried out by means of a computing program that calculates the eigenenergies and eigenvectors of the Hamiltonian in eqn. (1) for given values of Ji parameters. A subroutine calculates the magnetic susceptibility (χ) from the obtained eigenenergies and the Van Vleck expression (2) where E(0)i terms are the eigenenergies of the exchange Hamiltonian, E(1)i terms represent the Zeeman splittings and k is the Boltzmann constant. Second order magnetic energies E(2)i have been omitted as they are all zero in this theoretical treatment.
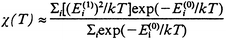
| (2) |
Fitting of the χmT experimental data was carried out by the usual least-squares procedure. Owing to the fact that the used least-squares function is not linear in the parameters, the minimisation was done by using the method of simulating annealing,19 followed and intercalated with single and Powell algorithms20 to obtain the absolute minimum value of the least-squares function. The error bars were obtained through variations of the individual parameters. Limits of the parameters were included in the Monte Carlo annealing taking into account the calculations made using Goodenough’s empirical rules, but they were wide enough to allow all permitted values for J positive and negative. Thus, this first approach did not lead to satisfactory fitting.
The fit can be improved by consideration of a non-isotropic Zeeman term in the Hamiltonian or a mean field correction, both implying the same bulk effect. Taking into consideration that the compound has nickel(II) cations, the anisotropy in the Zeeman term is thought to be caused by the zero-field-splitting (D). Thus, from the mathematical point of view, treatment of the influence of the D term on the spin levels for the tetramer is quite complicated. On the contrary, applying a mean field correction presents the advantage of being much easier to be quantified.
Obviously, the mean field correction for the compound accounts for the coupling between neighbouring tetramers. In this way, the interpretation of the magnetic properties can be carried out by considering both the intramolecular (Ji) and intermolecular interactions (zJ′ = enhancement parameter) on the basis of eqn. (3). According to eqn. (3), the best fit parameters were calculated to be J1 = +18.8(5), J2 = +6.9(3), J3 = +1.3(3), J4 = +0.2(1) cm−1, zJ′ = −0.6(2) cm−1 and g = 2.136(3). The corresponding theoretical curve (shown in Fig. 3) does reproduce both the region above and below the maximum at 12 K. This set of parameters indicates the occurrence of ferromagnetic intramolecular coupling along with antiferromagnetic intermolecular coupling. The exchange constants J and the g value lie between the expected ones for this type of compounds and will be compared below to some others found in the literature. These data should be interpreted on the basis of the intermolecular exchange especially affecting the magnetic behaviour at low temperatures. Thus, the fact that such a small zJ′ value clearly dominates the bulk susceptibility in this temperature region has previously been noticed in copper(II) systems.21
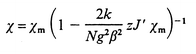
| (3) |
Fig. 4 shows the 19 energy levels which are thermally available for the compound (taking the Zeeman energy equal to zero and scaled for the energy of the ground state to be zero). As can be seen, the ground state corresponds to S = 4 which is in accordance with the ferromagnetic behaviour of the system (excluding the enhancement). The stabilisation of the highest spin state can be related to the existence of octahedra sharing edges, as suggested by Coronado and co-workers4a after studying the magnetic behaviour of Ni4 and Ni3 clusters. Thus, these researchers have also observed ferromagnetic–antiferromagnetic competition in Ni9 clusters that exhibit NiO6 octahedra sharing edges and corners.
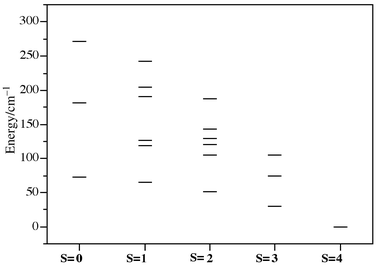
Table 4 shows the magnetic exchange constant values for dicubane-like tetramers. As mentioned above, the difference between our compound and the rest of them consists of the presence of Ni–(Nend-on azide,O)–Ni bridges in the former. Since all the interactions in the rest take place through Ni–(O,O)–Ni bridges, their magnetic properties have been interpreted on the basis of a two-J treatment (then J1 = J2 and J4 = 0). Thus, data in Table 4 clearly show that M–(O,O)–M bridges just provide ferromagnetic coupling for M = Ni. The latter can be explained if considering that, as reported by Ginsberg and co-workers,22 deviations of ±14° from 90° in Ni–O–Ni angles can be tolerated before the superexchange pathways lose their predominant ferromagnetic interaction. Thus, both nickel tetramers in Table 4, as well as some other nickel compounds reported elsewhere,4c,23 lie within this category. On the other hand, comparison between J values shows that the highest one (J1 = 18.8 cm−1 for our compound) corresponds, as expected, to the coupling through end-on azide.
Finally, it should be also noted that the J value obtained in this work for the coupling through Ni–(Nend-on azide,O)–Ni bridges (18.8 cm−1) is comparable to the value of 21.3 cm−1 reported by Ribas et al.5c for a nickel(II) tetramer also exhibiting Ni–(O,Nend-on azide)–Ni bridges.
Concluding remarks
The simultaneous use of dpk and azide has led to the preparation of a tetrameric nickel(II) cluster, which exhibits an unprecedented structural arrangement for metal(II)–azide systems. The structure consists of dicubane-like entities sharing a face with two missing vertexes where Ni–(O,Nend-on azide)–Ni and Ni–(O,O)–Ni bridges can be found. The magnetic characterisation is consistent with the occurrence of intramolecular ferromagnetic and intermolecular antiferromagnetic coupling. The calculated J values are the highest ones reported so far for similar dicubane compounds.Acknowledgements
This work has been carried out with the financial support of the Universidad del País Vasco/Euskal Herriko Unibertsitatea (Grant UPV 130310-EB201/1998), the Gobierno Vasco/Eusko Jaurlaritza (Project PI96/39) and the Ministerio de Educación y Cultura (Project PB97-0637). Z. E. S. also thanks the Ministerio de Educación y Ciencia for grant AP96 20174323. G. E. B. works for the ICTP (Trieste, Italy) Associated Scheme.References
- S. M. J. Aubin, M. W. Wemple, D. M. Adams, H.-L. Tsai, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 7746 CrossRef CAS.
- N. Mataga, Theor. Chim. Acta, 1968, 10, 378 CrossRef.
- S. Ferlay, T. Mallah, R. Ouahes, P. Veillet and M. Verdaguer, Inorg. Chem., 1999, 38, 229 CrossRef CAS; S. L. Castro, Z. Sun, C. M. Grant, J. C. Bellinger, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1998, 120, 2365 CrossRef CAS; V. Tangoulis, C. P. Raptopoulou, A. Terzis, E. G. Bakalbassis, E. Diamantopoulou and S. P. Perlepes, Inorg. Chem., 1998, 37, 3153 CrossRef CAS; G. Aromí, J.-P. Claude, M. J. Knapp, J. C. Huffman, D. N. Hendrickson and G. J. Christou, J Am. Chem. Soc., 1998, 120, 2977 CrossRef; M. W. Wemple, D. K. Coggin, J. B. Vincent, J. K. McCuster, W. E. Streib, J. C. Huffman, D. N. Hendrickson and G. J. Christou, J. Chem. Soc., Dalton Trans., 1998, 719 RSC; G. Aromí, S. M. J. Aubin, M. A. Bolcar, G. Christou, H. J. Eppley, K. Folting, D. N. Hendrickson, J. C. Huffmann, R. C. Squire, H.-L. Tsai, S. Wang and M. W. Wemple, Polyhedron, 1998, 17, 3005 CrossRef CAS; M. A. Bolcar, S. M. J. Aubin, K. Folting, D. N. Hendrickson and G. Christou, Chem. Commun., 1997, 1485 RSC; A. Caneschi, D. Gatteschi and R. J. Sessoli, J. Chem. Soc., Dalton Trans., 1997, 3963 RSC; D. P. Goldber, A. Caneschi, C. D. Delfs, R. Sessoli and S. J. Lippard, J. Am. Chem. Soc., 1995, 117, 5789 CrossRef CAS; A. K. Powell, S. L. Heath, D. Gatteschi, L. Pardi, R. Sessoli, G. Spina, F. D. Giallo and F. Pieralli, J. Am. Chem. Soc., 1995, 117, 2491 CrossRef CAS; R. Sessoli, H.-L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS.
- (a) J. M. Clemente-Juan, E. Coronado, J. R. Galán-Mascarós and C. J. Gómez-García, Inorg. Chem., 1999, 38, 55 CrossRef; (b) J. Faus, F. Lloret, M. Julve, J. M. Clemente-Juan, M. C. Muñoz and X. Solans and M. Font-Bardía, Angew. Chem., Int. Ed. Engl., 1996, 35, 1485 CrossRef CAS; (c) M. Salah El Fallah, E. Rentschler, A. Caneschi, R. Sessoli and D. Gatteschi, Inorg. Chem., 1996, 35, 3723 CrossRef; (d) A. J. Blake, E. K. Brechin, A. Codron, R. O. Gould, C. M. Grant, S. Parsons, J. M. Rawson and R. E. P. Winpenny, J. Chem. Soc., Chem. Commun., 1995, 1983 RSC; (e) E. G. Bakalbassis, E. Diamantopoulou, S. P. Perlepes, C. P. Raptopoulou, V. Tangoulis, A. Terzis and T. F. Zafiropoulos, J. Chem. Soc., Chem. Commun., 1995, 1347 RSC.
- (a) M. A. Halcrow, J.-S. Sun, J. C. Huffman and G. Christou, Inorg. Chem., 1995, 34, 4167 CrossRef; (b) M. A. Halcrow and J. C. Huffman and G. Christou, Angew. Chem., Int. Ed. Engl., 1995, 34, 889 CrossRef CAS; (c) J. Ribas, M. Monfort and X. Costa and X. Solans, Inorg. Chem., 1993, 32, 695 CrossRef CAS.
- J. A. R. Navarro, M. A. Romero, M. J. Salas, M. Quirós and E. R. T. Tiekink, Inorg. Chem., 1997, 36, 4988 CrossRef CAS; M. C. Feller and R. Robson, Aust. J. Chem., 1968, 21, 2919 CAS; A. Basu, T. G. Kasar and N. Y. Sapre, Inorg. Chem., 1998, 27, 4539; A. Basu, A. R. Saple and N. Y. Sapre, J. Chem. Soc., Dalton Trans., 1987, 1797 RSC; B. A. Bovikin , A. M. Omel’Chenko , R. N. Sharonina and I. A. Zanina , Abstracts of Proceedings of the 15th All-Union Chugaevskii Conference on the Chemistry of Complex Compounds, Ukraine, 1985, vol. 1, p. 229; Search PubMed; N. Okabe and M. Koizumi, Acta Crystallogr., Sect. C, 1998, 54, 288 Search PubMed; R. Faure and H. Loiseleur and G. Thomas-David, Acta Crystallogr., Sect. B, 1973, 29, 1890 CrossRef; S. L. Wang, J. W. Richardson, S. J. Brigg, R. A. Jacobson and W. P. Jensen, Inorg. Chim. Acta, 1986, 111, 67 CrossRef CAS; Z. Zerna, M. G. Barandika, R. Cortés, M. K. Urtiaga and M. I. Arriortua, Polyhedron, 1998, 18, 249 CrossRef CAS.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A, 1983, 39, 158 CrossRef.
- G. M. Sheldrick , SHELXS 97, Program for the Solution of Crystal Structures, University of Göttingen, 1997. Search PubMed.
- G. M. Sheldrick , SHELXL 97, Program for the Refinement of Crystal Structures, University of Göttingen, 1997. Search PubMed.
- International Tables for X-Ray Crystallography, Kynoch Press, Birmingham, 1974, vol. IV. Search PubMed.
- C. K. Johnson , ORTEP II, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976. Search PubMed.
- (a) S. R. Breeze, S. Wang, J. E. Creedan and N. P. Raju, J. Chem. Soc., Dalton Trans., 1998, 2327 RSC; (b) Y. Sumatsuki, H. Shimada, T. Matsuo, M. Nakamura, F. Kai, N. Matsumoto and N. Re, Inorg. Chem., 1998, 37, 5566 CrossRef; (c) H. Li, Z. J. Zhong and W. Chen and X.-Z. Tou, J. Chem. Soc., Dalton Trans., 1997, 463 RSC.
- K. Nakamoto , Infrared Spectra of Inorganic and Co-ordination Compounds, Wiley, New York, 1997. Search PubMed.
- R. R. Osborne and W. R. McWhinnie, J. Chem. Soc. A, 1967, 2075 RSC.
- F. E. Mabbs and D. J. Machin , Magnetism and Transition Metal Complexes, Chapman and Hall, London, 1973. Search PubMed.
- J. K. Kambé, J. Phys. Soc. Jpn., 1950, 5, 48 Search PubMed; W. E. Hatfield , in Theory and Applications of Molecular Paramagnetism, E. A. Bourdeaux and L. N. Mulay, Wiley-Interscience, New York, 1976, ch. 7. Search PubMed.
- J. B. Goodenough, Phys. Rev., 1965, 100, 564 Search PubMed; Phys. Chem. Solids, 1958, 6, 287. Search PubMed; J. Kanamori, Phys. Chem. Solids, 1959, 10, 87 Search PubMed.
- M. Rotenberg , R. Bivens , N. Metropolis and J. K. Wooten, Jr , The 3-j and 6-j symbols, Technology, MIT, Cambridge, MA, 1959. Search PubMed.
- S. Kirpatrick, C. D. Gelatt, Jr and M. P. Vecchi, Science, 1983, 220, 271 and refs. therein..
- J. A. Nelder and R. Mead, Comput. J., 1965, 7, 308 Search PubMed; R. P. Brent , Algorithms for minimization without derivatives, Prentice-Hall, New Jersey, 1973. Search PubMed.
- A. N. Papadopoulus, V. Tangoulis, C. P. Raptopoulou, A. Terzis and D. P. Kessiglou, Inorg. Chem., 1996, 35, 559 CrossRef and refs. therein..
- J. A. Bertrand, A. P. Ginsberg, R. I. Kaplan, C. E. Kirkwood, R. L. Martin and R. C. Sherwood, Inorg. Chem., 1971, 10, 240 CrossRef.
- A. Cornia, A. C. Fabretti, D. Gatteschi, G. Pálui, E. Rentschler, O. Shchegolikhina and A. A. Zhdanov, Inorg. Chem., 1995, 34, 5383 CrossRef CAS.
Footnote |
| † Supplementary data available: rotatable 3-D crystal structure diagram in CHIME format. See http://www.rsc.org/suppdata/dt/a9/a905430h/ |
| This journal is © The Royal Society of Chemistry 2000 |