Solvent-free oligomerization of phenylacetylene catalyzed by (cyclopentadienyl)nickel complexes
William E.
Douglas
CNRS UMR 5637, Case 007, Université de Montpellier II, 34095, Montpellier Cedex 5, France
First published on 27th January 2000
Abstract
The solvent-free reaction of phenylacetylene at 115 °C in the presence of nickelocene, [(η-Cp)Ni]2(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), [(η-Cp)Ni(CO)]2, (η-Cp)Ni(NO), (η-Cp)Ni(GeBr3)(CO), (η-Cp)Ni[(P(OMe3)]Cl, (η-Cp)Ni(Ph3P)Cl, (η-Cp)Ni(Bun3P)I, or (Ph3P)2Ni(CO)2 gives rise to a mixture of cyclotrimers, linear oligomers and poly(phenylacetylene), no reaction being observed in the case of internal acetylenes. Cyclotrimer formation is favoured by the presence of (a) added phosphine (2 equiv.), or (b) (cyclopentadienyl)nickel catalysts bearing a chloro substituent at Ni. A reduction in reaction temperature results in lower conversion but favours linear oligomer and polymer formation. The extent of reaction is greatly reduced in the case of (a) nickelocene in the presence of 2 equiv. PBun3, (b) (η-Cp)Ni(GeBr3)(CO), or (c) (η-Cp)Ni(NO). The main effect of the presence of solvent, regardless of whether it is potentially coordinating (toluene) or not (n-octane), is to suppress almost completely reactions catalyzed by nickelocene.
CH), [(η-Cp)Ni(CO)]2, (η-Cp)Ni(NO), (η-Cp)Ni(GeBr3)(CO), (η-Cp)Ni[(P(OMe3)]Cl, (η-Cp)Ni(Ph3P)Cl, (η-Cp)Ni(Bun3P)I, or (Ph3P)2Ni(CO)2 gives rise to a mixture of cyclotrimers, linear oligomers and poly(phenylacetylene), no reaction being observed in the case of internal acetylenes. Cyclotrimer formation is favoured by the presence of (a) added phosphine (2 equiv.), or (b) (cyclopentadienyl)nickel catalysts bearing a chloro substituent at Ni. A reduction in reaction temperature results in lower conversion but favours linear oligomer and polymer formation. The extent of reaction is greatly reduced in the case of (a) nickelocene in the presence of 2 equiv. PBun3, (b) (η-Cp)Ni(GeBr3)(CO), or (c) (η-Cp)Ni(NO). The main effect of the presence of solvent, regardless of whether it is potentially coordinating (toluene) or not (n-octane), is to suppress almost completely reactions catalyzed by nickelocene.
Introduction
During a previous study concerning the catalyzed cure of acetylene-terminated resins it was discovered that nickelocene is an effective catalyst for the polymerization of terminal acetylenes, but the reaction occurs only under solvent-free conditions.1 In the case of phenylacetylene the products consist of a mixture of cyclotrimers, linear oligomers and trans-cisoidal poly(phenylacetylene).2,3 Here are reported the results of an investigation into this reaction and its generalization to a variety of (cyclopentadienyl)nickel catalysts. It is shown that the nature and distribution of the products can be monitored by the use of size exclusion chromatography (SEC).Experimental
Phenylacetylene (Aldrich or Acros) was redistilled before use. The mass spectra were obtained by use of a JEOL JMS-DX300 instrument (EI or FAB). Elemental analyses [% Found (% Calculated)] were done at the CNRS Service Central d’Analyse. SEC was carried out in THF at a flow rate of 0.9 ml min−1 by use of a Waters 510 system equipped with 100, 500, 1000 and 10000 Å columns and a UV detector operating at 254 nm.Catalyst preparation/purification
The complex (PPh3)2Ni(CO)2 (Aldrich) was used as received, (η-Cp)Ni(PPh3)Cl (Aldrich) was recrystallized from CH2Cl2–pentane to give the pure compound [C 65.52 (65.53), H 4.90 (4.78)%], and nickelocene (Strem) was re-sublimed. Technical grade [(η-Cp)Ni(CO)]2 from Aldrich (containing much nickelocene) was fractionally vacuum-sublimed at 100 °C to give a small quantity (<20%) of fine dark-red crystals of the desired compound.The dimer [(η-Cp)Ni]2(PhCCH), prepared as described![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 4 from crude [(η-Cp)Ni(CO)]2, was isolated by chromatography followed by recrystallization from n-pentane at −78 °C to give dark-green crystals [mp 133–134 °C (blackening above 100 °C), lit.4 132–133 °C; M+ 348 (EI)].
4 from crude [(η-Cp)Ni(CO)]2, was isolated by chromatography followed by recrystallization from n-pentane at −78 °C to give dark-green crystals [mp 133–134 °C (blackening above 100 °C), lit.4 132–133 °C; M+ 348 (EI)].
(η-Cp)Ni(GeBr3)(CO) [M+ 464 (EI)] was prepared as described5 from pure [(η-Cp)Ni(CO)]2 in toluene. The complexes (η-Cp)Ni[P(OMe)3]Cl6 [M+ 282 (EI)], (η-Cp)Ni[P(n-Bu)3]I7 (mp 50–52 °C (waxy crystals formed over a period of 2 months), lit.8 red oil; M+ 452 [FAB: m-nitrobenzylalcohol]) and (η-Cp)Ni(NO)9 [M+ 153 (EI)] were prepared as described.
Preparation of tetraphenylcyclooctatetraene isomers
The previously described10 mixture of cyclooctatetraene isomers (in which no trace of trimers was detected)10 was prepared from phenylacetylene in the presence of chlorotrimethylsilane and Pd/C. The SEC trace of the crude reaction product showed only three peaks (retention times 37.90, 39.37 and 40.64 min).
11 as described12 and recrystallized from n-heptane (mp 101.1–101.6 °C, lit.13 two forms 100 °C and 119–120 °C). The IR spectrum showed no bands characteristic of linear trimers. A solution of KMnO4 in acetone was not decolourized by the compound thus confirming the absence of non-aromatic alkene groups.13 The SEC trace showed a single peak with a retention time of 42.03 min, identical to that for 1,3,5-triphenylbenzene (Aldrich). The SEC trace for a mixture of the two isomers also showed only a single peak with a retention time of 42.03 min.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C–CH
C–CH![[double bond, length half m-dash]](https://www.rsc.org/images/entities/b_char_e006.gif) CHPh..
β-Bromostyrene (ca. 85% trans and 15% cis) was coupled with phenylacetylene under the published conditions, Et3N being used in place of Et2NH.14 The SEC trace of the reaction product (containing mainly E-1,4-diphenylbut-1-en-3-yne with some Z-isomer14) showed a single peak with a retention time of 43.64 min. An identical trace was obtained after leaving the material to stand for 4 d.
C–CPhCH2..
The unstable15,16 branched enyne was prepared starting from α-bromostyrene under the same conditions as for the preceding reaction. After 6 h at RT the SEC trace for the reaction mixture showed a peak with a retention time of 43.64 min assigned to the compound together with a peak resulting from the monomer (retention time 46.20 min). There was also a barely perceptible peak with a retention time of 40.54 min. After 5 d, the magnitude of the peak for PhCC–CPh
CHPh..
β-Bromostyrene (ca. 85% trans and 15% cis) was coupled with phenylacetylene under the published conditions, Et3N being used in place of Et2NH.14 The SEC trace of the reaction product (containing mainly E-1,4-diphenylbut-1-en-3-yne with some Z-isomer14) showed a single peak with a retention time of 43.64 min. An identical trace was obtained after leaving the material to stand for 4 d.
C–CPhCH2..
The unstable15,16 branched enyne was prepared starting from α-bromostyrene under the same conditions as for the preceding reaction. After 6 h at RT the SEC trace for the reaction mixture showed a peak with a retention time of 43.64 min assigned to the compound together with a peak resulting from the monomer (retention time 46.20 min). There was also a barely perceptible peak with a retention time of 40.54 min. After 5 d, the magnitude of the peak for PhCC–CPh![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH2 (retention time 43.64 min) had decreased, the peak with a retention time of 40.54 min having become the most important (ratio of areas ca. 1∶2).
CH2 (retention time 43.64 min) had decreased, the peak with a retention time of 40.54 min having become the most important (ratio of areas ca. 1∶2).
Polymerization procedure
Unless otherwise stated (Tables 1 and 2), the catalyst (ca. 0.1 mol% [Ni]) was added to degassed phenylacetylene (0.930 g [1 ml]; 9.105 mmol) under dinitrogen. The stirred mixture was placed in a bath at 115 °C and the reaction tube was closed to prevent loss of material. In most cases there was a colour change to dark orange-red after a short period. The reaction mixture was cooled to RT after 6 h to give a dark red solid or viscous liquid. The SEC traces of the product mixtures thus obtained showed peaks resulting from some or all of the following: poly(phenylacetylene) (retention time ca. 32 min), linear oligomers of phenylacetylene (retention time ca. 40 min), 1,2,4- and/or 1,3,5-triphenylbenzene (retention time 42.03 min), unchanged phenylacetylene (retention time 45.00 min).a
| Composition of final reaction mixtureb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Reaction temperature/time (°C/h) | Solvent | Added ligand | Time for colour change to orange-red | Aspect of final reaction mixture at RT | Polymer (%)c |
Linear oligomers (%)d |
Cyclotrimers (%)e |
PhCCH (%)f |
Cyclotrimers: (polymer + linear oligomers) |
| a Nickelocene (ca. 0.01 mmol) and phenylacetylene (9.1 mmol). b Determined by SEC. c Retention time 31.3–36.0 min. d Retention time 39.3–40.3 min. e Retention time 42.03 min. f Retention time 45.00 min. | ||||||||||
| 1 | 115/6 | none | none | 5 min | dark orange-red solid | 50 | 9 | 30 | 8 | 1∶2.0 |
| 2 | 65/24 | none | none | 14 h | red-brown nonviscous liquid | 10 | 15 | 4 | 74 | 1∶6.3 |
| 3 | 100/7 | PhMe (5 ml) | none | 1 h | orange-red solution | 2.6 | 2.8 | 2.4 | 92 | 1∶2.3 |
| 4 | 100/7 | n-octane (5 ml) | none | 1 h | orange-red solution | 1.5 | 2.3 | 1.3 | 95 | 1∶2.9 |
| 5 | 115/6 | none | PBun3 (1 equiv./(η-Cp)2Ni) | 3 h | orange-red viscous liquid | 47 | 15 | 32 | 8 | 1∶1.9 |
| 6 | 115/6 | none | PBun3 (2 equiv./(η-Cp)2Ni) | 6 h | orange-red liquid | 17 | 12 | 24 | 46 | 1∶1.2 |
| 7 | 115/6 | none | PPh3 (2 equiv./(η-Cp)2Ni) | 1 h | orange-red viscous liquid | 8 | 7 | 77 | 1.5 | 1∶0.2 |
a
| Composition of final reaction mixtureb |
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Time until colour changed to orange-red | Aspect of final reaction mixture at RT | Polymer (%)c |
Linear oligomers (%)d |
Cyclotrimers (%)e |
PhCCH (%)f |
Cyclotrimers: (polymer + linear oligomers) |
| a Catalyst (ca. 0.01 mmol) and phenylacetylene (9.1 mmol) heated at 115 °C for 6 h. b Determined by SEC. c Retention time 31.83–32.22 min. d Retention time 39.19–40.31 min. e Retention time 42.03 min. f Retention time 45.00 min. g Reaction temperature 65 °C. | ||||||||
| 1 | [(η-Cp)Ni]2(PhCCH) |
2 min | dark orange-red solid | 53 | 7 | 30 | 8 | 1∶2.0 |
| 2 | [(η-Cp)Ni(CO)]2 | 30 sec | dark orange-red viscous liquid | 40 | 20 | 35 | 8 | 1∶1.7 |
| 3 | (η-Cp)NiNO | (red catalyst) | brown liquid | 23 (1g) |
2 (3g) |
11 (1g) |
65 (95g) |
1∶2.3 (1∶4.0g) |
| 4 | (η-Cp)Ni(GeBr3)(CO) | 1 min | orange-red liquid | 26 | 0 | 15 | 59 | 1∶1.7 |
| 5 | (η-Cp)Ni(PBun3)I | 1 h | orange-red viscous liquid | 56 | 8 | 29 | 8 | 1∶2.2 |
| 6 | (η-Cp)Ni[P(OMe)3]Cl | 1 min | orange-red viscous liquid | 38 | 15 | 40 | 3 | 1∶1.3 |
| 7 | (η-Cp)Ni(PPh3)Cl | (red catalyst) | red viscous liquid | 52 | 1 | 38 | 7 | 1∶1.4 |
| 8 | (PPh3)2Ni(CO)2 | immediate | dark orange-red solid | 35 | 0 | 60 | 6 | 1∶0.6 |
Results and discussion
The (Ph3P)2Ni(CO)2-catalyzed polymerization of acetylenes in solution was reported many years ago.17 With phenylacetylene in alcohol at 70 °C a violent reaction occurs giving a gum containing triphenylbenzene.13,18 The reaction in inert solvents such as refluxing cyclohexane has been extensively studied by Meriwether et al.19–22 In particular, the results of experiments with deuterated acetylenes are consistent with a hydrogen transfer step for both linear and cyclic polymerization of phenylacetylene, the suggested mechanism [Scheme 1 with (Ph3P)2 in place of (η-Cp)] accounting for both processes.22 Of the many disubstituted acetylenes investigated only 2-butyne-1,4-diol reacted.19 A disputed23 second mechanism involving a nickelole intermediate was put forward to explain the formation of the hexasubstituted cyclotrimer in this case,22 but it should be noted that 2-butyne-1,4-diol undergoes spontaneous dimerization and cyclotrimerization in protic solvents.24 The Meriwether mechanism has also been applied in the case of Ni(acac)2–AliBu3 for which the intermediate ethynyl hydride complex of Ni has been isolated.25
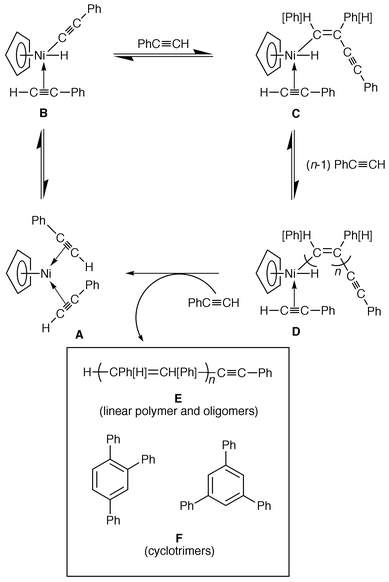 | ||
| Scheme 1 Reaction mechanism for nickelocene-catalyzed polymerization and cyclotrimerization of phenylacetylene in the absence of solvent. | ||
Previously, we have reported that under solvent-free conditions nickelocene catalyzes terminal-acetylene polymerization,1 in the case of phenylacetylene giving cyclotrimers and linear polyenes but no cyclotetramers.2,3 In solution, nickelocene, and also [(η-Cp)Ni]2(RCCR′), catalyze the polymerization of acetylenes but only in the presence of an aromatic heterocyclic amine (e.g. pyridine)26 or AlBr327 (nickelocene with F3CC CCF3 in solution has been reported to give traces of the cyclotrimer28).
Product SEC retention times
In order to investigate the feasibility of using SEC for monitoring the polymerization reaction, the polymerization of phenylacetylene in the presence of nickelocene was repeated under the same conditions as those used previously when the reaction products were separated and analyzed.3 The SEC trace showed four peaks (Fig. 1 and Table 1, entry 1) corresponding to the four components previously separated from the reaction mixture and characterized:3 (i) retention time 32.01 min; trans-cisoidal poly(phenylacetylene) (Mwca. 3000 [polydispersity 1.6] with respect to polystyrene standards), (ii) 40.30 min; linear oligomers of phenylacetylene (that with n = 4.5 has been previously isolated3), (iii) 42.03 min; 1,2,4- and 1,3,5-triphenylbenzene, and (iv)
45.00 min; unchanged phenylacetylene. However, there are some discrepancies between the relative amounts of each component determined by the two methods; isolated3 (found by SEC), (i) 39 (50), (ii) 30 (9), (iii) 24 (30), and (iv) 8 (8)%. These differences can be explained by variations in sensitivity of the UV detector, and also by the fact that some of the material isolated in the linear oligomer fraction is not included in the area measured for the SEC peak with a retention time of 40.30 min.
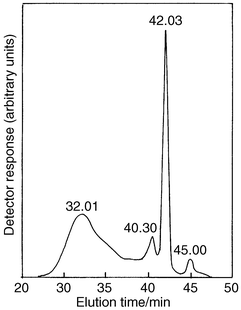 | ||
| Fig. 1 SEC trace for product mixture from reaction of phenylacetylene in the presence of nickelocene under the standard conditions (Table 1, entry 1). | ||
It having been established that the components in the reaction mixture could be distinguished by SEC, various other possible products were synthesized in order to check for any coincidence of retention times.
The mixture of tetraphenylcyclooctatetraene isomers (possibility of 7 isomers29), prepared in such a way as to contain no trace of cyclotrimers,10 showed three SEC peaks with retention times 37.90, 39.37 and 40.64 min. In particular, the peak at 37.90 min allows tetraphenylcyclooctatetraene formation in the reaction mixture to be distinguished by SEC (cf. Fig. 1 for the case where the absence of cyclotetramers has been established by other means3).
The SEC trace of pure 1,2,4-triphenylbenzene contained a single peak with a retention time of 42.03 min, identical to that for 1,3,5-triphenylbenzene, thus showing that the two cyclotrimer isomers cannot be distinguished by SEC.
The stable tail-to-tail dimer PhCC–CHCHPh was prepared from a commercial sample of β-bromostyrene (ca. 85% trans and 15% cis) by Pd-catalyzed coupling with phenylacetylene.14 The SEC trace of the reaction product (containing mainly E-1,4-diphenylbut-1-en-3-yne with some Z-isomer) showed a single peak with a retention time of 43.64 min. An identical trace was obtained after leaving the material to stand for 4 d. SEC can therefore be used to distinguish the presence of the dimer in the reaction mixture.
Starting from α-bromostyrene, the unstable15,16 head-to-tail dimer PhCC–CPhCH2 was prepared in the same way, the SEC trace of the product mixture showing peaks at 43.64 min for the head-to-tail dimer and at 46.20 min for unchanged monomer. There was also a barely perceptible peak with a retention time of 40.54 min. After 5 d, the magnitude of the peak for PhCC–CPhCH2 had decreased and the peak for the decomposition product at 40.54 min had become the most important (ratio of areas ca. 1∶2). The retention time of the decomposition product is consistent with it being a mixture of linear oligomers of phenylacetylene. Thus, SEC allows dimer formation to be distinguished in the reaction mixture but no differentiation between the tail-to-tail and head-to-tail isomers is possible.
The preliminary studies showed the feasibility of using SEC to follow the effects of catalysts on product distribution. Indeed, the absence of SEC peaks with retention times of ca. 37.9 min or ca. 43.6 min rules out any significant cyclotetramer or dimer formation. In all cases only the four products previously isolated and characterized for nickelocene are formed, their distribution being dependent on the nature of the nickel catalyst and reaction conditions.
Nickelocene-catalyzed polymerization of phenylacetylene
Treatment of phenylacetylene with nickelocene under the standard conditions (115 °C, 6 h) in the absence of solvent resulted in 92% conversion, the final reaction mixture containing 50% polymer (Table 1, entry 1). The initial deep green solution became rapidly olive-green and after 5 min the mixture was orange-red. The reaction was unaffected by the presence of the radical scavenger 2,6-di-tert-butyl-4-methylphenol. Lowering the reaction temperature to 65 °C for 24 h resulted in a much reduced degree of conversion (26%), the product mixture becoming poorer in cyclotrimers (Table 1, entry 2).No reaction of internal acetylenes (Me3SiCCSiMe3, PhC CSiMe3, PhCCPh) occurred under solvent-free conditions in the presence of nickelocene or the other (cyclopentadienyl)nickel catalysts. This suggests that, as with (Ph3P)2Ni(CO)2, the reaction mechanism includes an acetylene hydrogen transfer step rather than involving nickelole or other such intermediates.30,31 Probably the mechanism operating in this case is that of Meriwether (Scheme 1), the catalytic species A being formed by loss of a cyclopentadienyl ring, a reaction which is known to occur readily.32,33 Indeed, a variety of 19-electron (η-Cp)NiL2 complexes, including (η-Cp)NiPPh3(HCCPh) and (η-Cp)Ni(HCCPh)2, have been studied in benzene
solution and are surprisingly stable.34 The following step in the catalytic cycle is the oxidative addition of phenylacetylene giving B, such intermediate ethynyl hydride complexes of Ni having been previously isolated.25 The next step is the insertion of phenylacetylene into the Ni–C bond, a reaction which has been previously observed.35–37 This step is repeated (n − 1)-fold giving D which re-forms A by reductive elimination of linear polymer E. The cyclotrimers F can be formed by reductive elimination from D when n = 2.
In order to investigate the effect of solvents, the nickelocene-catalyzed reaction was carried out at 100 °C for 7 h in the presence of 5 ml of toluene or n-octane, the concentration of the phenylacetylene solution being ca. 17% v/v. In each case, the mixture became yellow almost immediately and after 1 h orange-red. Evaporation of the reaction mixtures gave traces of reddish-brown oils, the weights obtained corresponding to 8% and 5% overall reaction (Table 1, entries 3 and 4), respectively. The main effect of the presence of solvent, regardless of whether it is potentially coordinating (toluene) or not (n-octane), is to suppress almost completely reactions catalyzed by nickelocene. In the case of nickelocene, coordination of a solvent molecule occurs at the expense of an acetylene molecule in the 19-electron catalytic species A (Scheme 1) thus inhibiting the reaction. This is in contrast to (Ph3P)2Ni(CO)2 where the corresponding catalytic species is 18-electron and can coordinate a molecule of solvent in addition to the acetylene molecules.
The presence of 1 equiv. of PBun3 (high electron-donating ability and moderate cone angle38) per (η-Cp)2Ni (Table 1, entry 5) had little effect. In contrast, with 2 equiv. PBun3 per (η-Cp)2Ni the extent of conversion fell to 54% (Table 1, entry 6). This was accompanied by an increase in the proportion of cyclotrimers suggesting that the phosphine is present in the coordination sphere of the catalytic species. Previously it has been found that the electronic and steric factors of the phosphorus ligand play an important rôle in product distribution in the solution oligomerization of phenylacetylene catalyzed by rhodium(I) complexes,39,40 the presence of excess electron-donating phosphites
inhibiting the reaction.40 Indeed, the sterically-hindered complex (tBu2PC2H4PtBu2)Ni(C2H2) does not catalyze cyclooligomerization or polymerization of acetylenes.41
The addition of 2 equiv. PPh3 (moderate electron-donating ability and large cone angle38) per (η-Cp)2Ni had a very different result, the extent of reaction being almost complete (98.5%) with selective formation of cyclotrimers in 77% yield (Table 1, entry 7). Presumably, triphenylphosphine is present in the coordination sphere of Ni replacing one acetylene group, thus favouring reductive elimination of the cyclotrimers. Nickelocene reacts with PPh3 in cyclohexane to give Ni(PPh3)4 quantitatively,42 but (η-Cp)Ni(PPh3)Cl and CpCl are formed in the presence of CCl4.32,33 Analogous reactions occur in the case of phosphites.43 The intermediate species (η-Cp)Ni(PPh3)(σ-Cp) may be formed by a π to σ rearrangement
of a cyclopentadienyl ligand.32,44 In the presence of excess phosphine, ionic species such as [(η-Cp)Ni(PR3)2]+Cl− are formed.45,46 Alkyl compounds of the type (η-Cp)Ni(PPh3)R are sufficiently stable to be isolated and characterized, an investigation of the n-alkyl derivatives suggesting reversible olefin β-elimination accompanied by nickel hydride formation.47 Nineteen-electron complexes (η-Cp)Ni(PR3)2 have been isolated and characterized, and have been shown to be most probably intermediates in the reactions of nickelocene with phosphines.48
Solvent-free polymerization in the presence of other (cyclopentadienyl)nickel catalysts
Use of the green phenylacetylene-bridged binuclear compound [(η-Cp)Ni]2(PhCCH) or the carbonyl-bridged binuclear complex [(η-Cp)Ni(CO)]2 (Table 2, entries 1 and 2) gave results very similar to those with nickelocene. In the case of [(η-Cp)Ni(CO)]2 most probably the known reaction4 to give [(η-Cp)Ni]2(PhCCH) takes place initially. Indeed, when the internal acetylene PhCCPh was treated with 0.35 mol% [(η-Cp)Ni(CO)]2 at 115 °C in the absence of solvent, the initially red solution turned deep green within 2 min consistent with formation of [(η-Cp)Ni]2(PhCCPh) (no oligomerization of the acetylene was observed). The kinetics of the reaction of [(η-Cp)Ni(CO)]2 with acetylenes to give [(η-Cp)Ni]2(RCCR′) have been extensively investigated,49,50
and it was found that at very high acetylene concentrations a second order mechanism is important.50 Green acetylene-bridged binuclear compounds are readily formed on reaction of acetylenes with nickelocene,51 and the complexes are stable up to 70–80 °C in an inert solvent.52 Most probably the nickelocene-catalyzed reaction involves an initial conversion step to give [(η-Cp)Ni]2(PhCCH) which then subsequently forms the catalytic species A (Scheme 1). This is consistent with the shorter times for the colour-change to orange-red in the case of [(η-Cp)Ni]2(PhCCH) or [(η-Cp)Ni(CO)]2 the binuclear intermediates being present initially in the reaction mixture.
The complex (η-Cp)Ni(NO) gives only 35% conversion at 115 °C. Decrease in the reaction temperature to 65 °C results in a reduction in both the extent of reaction (to 5%) and, as with nickelocene, the proportion of cyclotrimers (Table 2, entry 3). Loss of the NO ligand enabling formation of [(η-Cp)Ni]2(PhC CH) occurs only with difficulty. Indeed, it has been found that nitrosyl exchange with 15NO did not occur for (η-Cp)Ni(NO) in 10 d at 120 °C.53
The complex (η-Cp)Ni(GeBr3)(CO) gave very similar results (Table 2, entry 4) to those for (η-Cp)Ni(NO), suggesting that although the CO ligand is readily lost (as shown by the initially green solution turning orange-red after only 1 min at 115 °C), the presence of the electron-withdrawing GeBr3 substituent5 hinders subsequent reactions with phenylacetylene. The reaction mechanism may involve 20-electron species (Scheme 1 with (η-Cp)(GeBr3)Ni in place of (η-Cp)Ni). No reaction was observed with PhCCPh.
In the case of (η-Cp)Ni(Bun3P)I, the product distribution and extent of reaction (Table 2, entry 5) were very similar to those for nickelocene (Table 1, entry 1), but the reaction mixture did not pass through a green stage. Here too the reaction mechanism may involve 20-electron species [Scheme 1 with (η-Cp)NiI in place of (η-Cp)Ni].
The complexes (η-Cp)Ni[(P(OMe3)]Cl and (η-Cp)Ni(PPh3)Cl (Table 2, entries 6 and 7) gave very similar results, the product mixtures being much richer in cyclotrimers than in the case of nickelocene. Since the phosphorus ligands are quite different in nature,38 the phosphine groups are probably not present in the active catalytic species. The reaction mechanism in each case may involve 20-electron species [Scheme 1 with (η-Cp)NiCl in place of (η-Cp)Ni]. Indeed, in neither case was a green stage observed corresponding to formation of [(η-Cp)Ni]2(PhCCH). This is in contrast to the case of nickelocene in the presence of 2 equiv. PPh3 (Table 1, entry 7) where cyclotrimer formation is favoured, the phosphine presumably playing a rôle in the co-ordination sphere of Ni (vide supra).
Finally, for comparison, the effect of (Ph3P)2Ni(CO)2 was investigated in the absence of solvent (Table 2, entry 8). Unlike the reaction in benzene where only cyclotrimer and linear trimer are afforded,19 the solvent-free reaction of phenylacetylene in the presence of (Ph3P)2Ni(CO)2 gives linear polymer in addition to cyclotrimer consistent with the much greater concentration of phenylacetylene favouring a larger value of n in intermediate D [Scheme 1 with (Ph3P)2 in place of (η-Cp)]. No reaction was observed with PhCCPh.
In summary, the oligomerization and cyclotrimerization of phenylacetylene with high conversion is catalyzed under solvent-free conditions by a wide variety of cyclopentadienylnickel complexes, internal acetylenes being unreactive. Cyclotrimer formation is favoured by the presence of (a) 2 equiv. of phosphine in the reaction mixture, or (b) (cyclopentadienyl)nickel catalysts bearing a chloro substituent at Ni. A reduction in reaction temperature results in lower conversion but favours linear oligomer and polymer formation. The extent of reaction is greatly reduced in the case of (a) (η-Cp)Ni(NO), (b) (η-Cp)Ni(GeBr3)(CO), or (c) nickelocene in the presence of 2 equiv. PBun3. The main effect of the presence of solvent, regardless of whether it is potentially coordinating (toluene) or not (n-octane), is to suppress almost completely reactions catalyzed by nickelocene. It can be concluded that under solvent-free conditions (cyclopentadienyl)nickel compounds in general are active catalysts for the polymerization of terminal acetylenes.
References
- W. Douglas and A. Overend, J. Organomet. Chem., 1986, 308, C14 CrossRef CAS.
- W. E. Douglas and A. S. Overend, J. Organomet. Chem., 1993, 444, C62 CrossRef CAS.
- W. E. Douglas and A. S. Overend, J. Mater. Chem., 1994, 4, 1167 RSC.
- J. F. Tilney-Bassett, J. Chem. Soc., 1961, 577 RSC.
- R. C. Edmondson, E. Eisner, M. J. Newlands and L. K. Thompson, J. Organomet. Chem., 1972, 35, 119 CrossRef CAS.
- J. Clemens, H. Neukomm and H. Werner, Helv. Chim. Acta, 1974, 57, 2000 CrossRef CAS.
- N. Kuhn and M. Winter, J. Organomet. Chem., 1984, 269, C47 CrossRef CAS.
- F. Matthey, J. Organomet. Chem., 1975, 87, 371 CrossRef.
- R. B. King , in Organometallic Syntheses, eds. J. J. Eisch and R. B. King, Academic Press, New York, 1965, vol. 1, pp. 169–171 Search PubMed.
- A. K. Jhingan and W. F. Maier, J. Org. Chem., 1987, 52, 1161 CrossRef CAS.
- S. V. Dighe and M. Orchin, Inorg. Chem., 1962, 1, 965 CrossRef CAS.
- W. Hübel and C. Hoogzand, Chem. Ber., 1960, 103, 93 Search PubMed.
- C. G. Overberger and J. M. Whelan, J. Org. Chem., 1959, 24, 1155 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467 CrossRef CAS.
- L. Carlton and G. Read, J. Chem. Soc., Perkin Trans. 1, 1978, 1631 RSC.
- J. Ohshita, K. Furumori, A. Matsuguchi and M. Ishikawa, J. Org. Chem., 1990, 55, 3277 CrossRef CAS.
- W. Reppe, Ann., 1948, 560, 104 Search PubMed.
- J. D. Rose and F. S. Statham, J. Chem. Soc., 1950, 69 RSC.
- L. S. Meriwether, E. C. Colthup, G. W. Kennerly and R. N. Reusch, J. Org. Chem., 1961, 26, 5155 CrossRef CAS.
- L. S. Meriwether, E. C. Colthup and G. W. Kennerly, J. Org. Chem., 1961, 26, 5163 CAS.
- E. C. Colthup and L. S. Meriwether, J. Org. Chem., 1961, 26, 5169 CAS.
- L. S. Meriwether, M. F. Leto, E. C. Colthup and G. W. Kennerly, J. Org. Chem., 1962, 27, 3930 CrossRef CAS.
- V. O. Reikhsfel’d and K. L. Makovetskii, Usp. Khim., 1966, 35, 1204 Search PubMed; Russ. Chem. Rev. (Engl. Transl.), 1966, 35, 510 Search PubMed.
- M. d’Amboise, D. Mathieu and D. L. Piron, Talanta, 1988, 35, 763 CrossRef CAS.
- G. A. Chukhadzhyan, Z. I. Abramyan and G. A. Gevorkyan, Zh. Obshch. Khim., 1973, 43, 2012 Search PubMed; J. General Chem. USSR (Engl. Trans.), 1973, 43, 1998 Search PubMed.
- M. Dubeckand A. H. Filbey , US Pat., 3256260, 1966(Chem. Abstr., 1966, 65, 7307a.) Search PubMed.
- V. O. Reikhsfel’d, B. I. Lein and K. L. Makovetskii, Dokl. Akad. Nauk SSSR, 1970, 190, 125 ( (Proc. Acad. Sci. USSR , 1970 , 190 , 7307a) ) Search PubMed.
- J. L. Davidson and D. W. A. Sharp, J. Chem. Soc., Dalton Trans., 1976, 1123 RSC.
- R. Diercks, L. Stamp and H. tom Dieck, Chem. Ber., 1984, 117, 1913 Search PubMed.
- R. E. Colborn and K. P. C. Vollhardt, J. Am. Chem. Soc., 1986, 108, 5470 CrossRef CAS.
- J. J. Eisch, J. E. Galle, A. A. Aradi and M. P. Boleslawski, J. Organomet. Chem., 1986, 312, 399 CrossRef CAS.
- Y. T. Ustynyuk, T. I. Voevodskaya, N. A. Zharikova and N. A. Ustynyuk, Dokl. Akad. Nauk SSSR, 1968, 181, 372 Search PubMed; Dokl. Chem. (Engl. Transl.), 1968, 181, 640 Search PubMed.
- C. Moberg and M. Nilsson, J. Organomet. Chem., 1973, 49, 243 CrossRef CAS.
- A. Becalska, J. D. Debad, H. K. Sanati and R. H. Hill, Polyhedron, 1990, 9, 581 CrossRef CAS.
- J. M. Huggins and R. G. Bergman, J. Am. Chem. Soc., 1979, 101, 4410 CrossRef CAS.
- S. J. Tremont and R. G. Bergman, J. Organomet. Chem., 1977, 140, C12 CrossRef CAS.
- L. M. Zubritskii, T. N. Fomina, V. A. Kalinina and K. V. Bal’yan, Zh. Org. Khim., 1979, 15, 2213 Search PubMed; J. Org. Chem. USSR (Engl. Transl.), 1979, 15, 2006 Search PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- S. Yoshikawa, J. Kiji and J. Furukawa, Makromol. Chem., 1977, 178, 1077 Search PubMed.
- J. Wendt, U. Klinger and H. Singer, Inorg. Chim. Acta, 1991, 183, 133 CrossRef CAS.
- K.-R. Pörschke, C. Pluta, B. Proft, F. Lutz and C. Krüger, Z. Naturforsch., Teil B, 1993, 48, 608 Search PubMed.
- H. Behrens and K. Meyer, Z. Naturforsch., Teil B, 1966, 21, 489 Search PubMed.
- V. Harder and H. Werner, Helv. Chim. Acta, 1973, 56, 1620 CrossRef CAS.
- C. Moberg, J. Organomet. Chem., 1976, 108, 125 CrossRef CAS.
- M. Sato, F. Sato and T. Yoshida, J. Organomet. Chem., 1971, 26, C49 CrossRef CAS.
- M. Sato, F. Sato and T. Yoshida, J. Organomet. Chem., 1971, 27, 273 CrossRef CAS.
- J. Thomson and M. C. Baird, Can. J. Chem., 1970, 48, 3443 CAS.
- E. K. Barefield, D. A. Krost, D. S. Edwards, D. G. van Derveer, R. L. Trytko and S. P. O’Rear, J. Am. Chem. Soc., 1981, 103, 6219 CrossRef CAS.
- P. C. Ellgen, Inorg. Chem., 1972, 11, 2279 CrossRef CAS.
- P. L. Stanghellini, R. Rossetti, O. Gambino and G. Cetini, Inorg. Chim. Acta, 1974, 10, 5 CrossRef CAS.
- H. Brunner and W. Pieronczyk, Bull. Soc. Chim. Belg., 1977, 86, 725 Search PubMed.
- E. W. Randall, E. Rosenberg, L. Milone, R. Rossetti and P. L. Stanghellini, J. Organomet. Chem., 1974, 64, 271 CrossRef CAS.
- F. A. Palocsay and J. V. Rund, Inorg. Chem., 1969, 8, 696 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |