DOI:
10.1039/A903712H
(Review Article)
Chem. Soc. Rev., 2000,
29, 57-67
The guanidine metabolites of Ptilocaulis
spiculifer and related compounds; isolation and synthesis
Received 10th May 1999
First published on UnassignedUnassigned11th January 2000
Abstract
Marine natural products possessing guanidine functionalities
display a considerable array of biological activity and not surprisingly
have attracted considerable synthetic interest. This review discusses the
isolation of several guanidine containing metabolites, primarily from the
sponge Ptilocaulis spiculifer, but also from other marine
organisms. It also explores the synthetic methodologies adopted for their
preparation and speculates on the structural similarity of the metabolite
ptilomycalin A to abiotic guanidine based anionic receptor molecules.
 Laura Heys Laura Heys | Laura Heys graduated (MChem) from Bangor University in 1999,
she is currently pursuing a career in the chemical industry. |
 Christopher G. Moore Christopher G. Moore | Christopher G. Moore graduated (BSc Hons) from Bangor in 1995
and is currently finishing his postgraduate studies which have focused on
the total synthesis of guanidine containing marine
metabolites. |
 Patrick J. Murphy Patrick J. Murphy | Patrick J. Murphy graduated (BSc Hons) from the University of
Manchester Institute of Science and Technology in 1983 and went on to
complete a PhD entitled ‘Synthetic routes towards naturally occurring
thiotetronic acids’ in 1986 under the supervision of Dr John Brennan
at the same institute. After a period of postdoctoral research at the
University of Salford under the supervision of Professor Garry Procter
(organosilicon chemistry) and Dr Mike Casey (reactions of chiral
sulfoxides), he was appointed to the post of Lecturer in Organic Chemistry
at the University of Wales, Bangor, gaining promotion to Senior Lecturer in
1999. Since gaining this post he has concentrated his research efforts on
the development of new methodology and its applications in synthesis, with
two key themes being the synthesis of naturally occurring guanidine
containing marine alkaloids and new applications of Wittig
chemistry. |
1 Isolation of ptilomycalin A and related
metabolites
Naturally occurring and synthetic compounds containing guanidine are of
considerable interest due both to the hydrogen-bond mediated interaction of
guanidinium ions and because of the wide range of biological activities
these substances display.1 Recent interest
in marine natural products has seen a steady increase in the number of
metabolites isolated from these sources and a metabolite of particular
interest isolated from the sponge Ptilocaulis spiculifer2 in 1989 is ptilomycalin A 1. This
alkaloid has a unique and fascinating structure consisting of a pentacyclic
guanidinium core linked to a spermidine unit via a ω-hydroxy
acid spacer group. The same compound was also isolated from a Red Sea
sponge of the genus Hemimycale sp.3
and subsequently from the sponge Batzella sp.4 and the starfishes Fromia monilis and
Celerina heffernani5 (Scheme 1).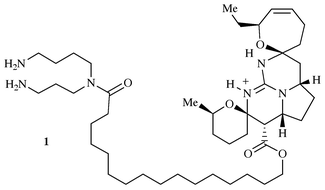 |
| | Scheme 1 | |
Ptilomycalin A displays a remarkable range of biological activities
including cytotoxicity against the following cell lines; P388
(IC50 0.1 μg mL−1), L1210 (IC50
0.4 μg mL−1), and KB (IC50 1.3 μg
mL−1) in addition to antifungal activity against
Candida albicans (MIC 0.8 μg mL−1) as well as
very good antiviral activity (HSV) at a concentration of 0.2 μg
mL−1.2
Previous to the isolation of ptilomycalin A the related guanidinium
alkaloids ptilocaulin 2 and isoptilocaulin 3 had been
isolated from the same sponge in 19816
(Scheme 2).
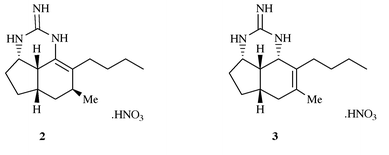 |
| | Scheme 2 | |
Several metabolites related to ptilomycalin A have subsequently been
isolated including the crambescidins 800, 816, 830 and 844
(4–7) from the Mediterranean sponge Crambe
crambe7 and in the case of 4
also from the sponge Monanchora arbuscula.8 The structures of these closely related guanidines
were elucidated in 1991 and differ from ptilomycalin A by the presence of a
hydroxyspermidine instead of a spermidine residue, the presence of an extra
hydroxy-substituent on C-13 (in the pyrrolidine ring) in the case of
crambescidins 816, 830 and 844 and in the variation in length of the
ω-hydroxy fatty acid. These polycyclic guanidines were reported to
exhibit antiviral activity against Herpes simplex virus type 1
(HSV-1) and they were cytotoxic to L1210 murine leukaemia cells7 and crambescidin 816 has been shown to be a potent
calcium channel blocker.9 In addition two
related guanidinium alkaloids were isolated from the New Caledonian
starfishes Celerina heffernani and Fromia monilis, and
named celeromycalin 8, which bears an extra hydroxy function on
the ω-hydroxy fatty acid and fromiamycalin 9 which contained
a modified spermidine unit (Scheme
3).
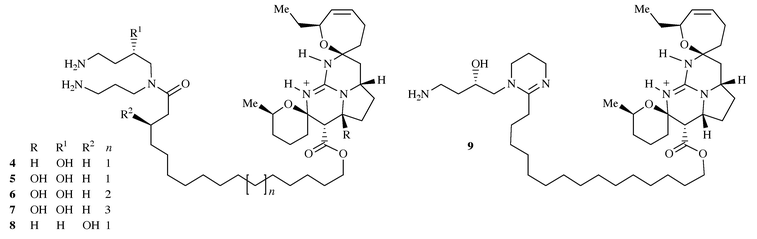 |
| | Scheme 3 | |
Interestingly the polar extracts of the starfish Fromia monilis
also contained ptilomycalin A and crambescidin 800, which allows us to
speculate that these metabolites may have arisen from a symbiotic organism
common to both species. Both celeromycalin and fromiamycalin showed
antiviral activities against the Herpes simplex virus (HSV-1) and
were also found to be highly cytotoxic in anti-HIV assays on CEM 4 cells
infected with human immunodeficiency virus (HIV-1) with CC-50 (cytotoxic
concentration) of 0.11 μg mL−1 (ptilomycalin A,
crambescidin 800 and fromiamycalin) and 0.32 μg mL−1
(celeromycalin).5
Possibly one of the most interesting of these metabolites to be isolated
is 13,14,15-isocrambescidin 800 10, the structure of which was
shown to contain a trans-ring junction in the guanidine
pentacycle.3,10 Surprisingly this
molecule is substantially less cytotoxic to L1210 cells than the other
crambescidins, and has no observed antiviral activity against HSV-1. This
observation might suggest that the enclosed ionic pocket found in
ptilomycalin A and the crambescidins and lacking in 10 might be
conferring much of the biological activity found in these compounds
(Scheme 4).
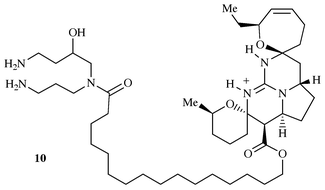 |
| | Scheme 4 | |
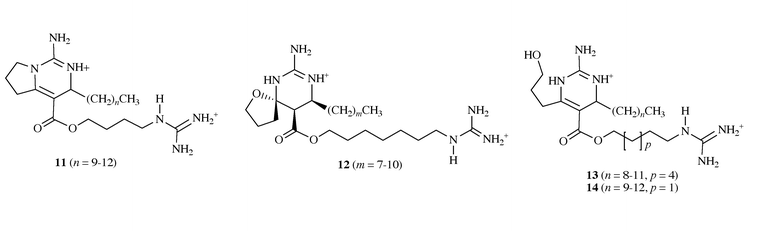
In addition, a series of related guanidine alkaloids, the crambescins A,
B, C1 and C2 11–14, were isolated from the same
sponge Crambe crambe and displayed cytotoxicity against L1210
cells (IC50 of less than 1 μg mL−1) and were
found to be ichthyotoxic (toxic against fish)11 (Scheme
5).
 |
| | Scheme 5 | |
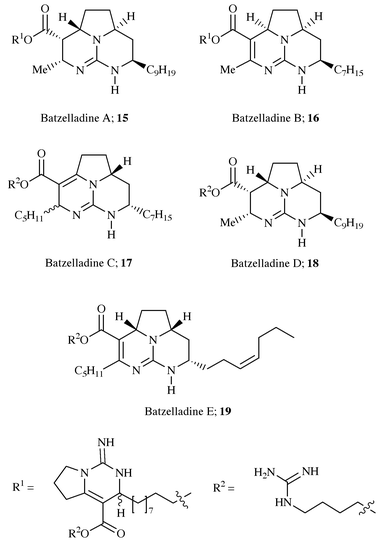
The batzelladines A–E 15–19, a series of
guanidine alkaloids containing a tricyclic guanidine unit, were isolated
from the Caribbean sponge Batzella sp. which was also found to
contain the previously isolated metabolites ptilomycalin A, ptilocaulin,
crambescin A, crambescidin 800 and 816.4,12 Batzelladines A and B were reported to inhibit the
binding of HIV-gp120 to the CD4 cell-surface receptor protein on T cells
and to be of interest in the treatment of AIDS.4 The structures of the batzelladines A–E were
originally elucidated by interpretation of the spectral data of the natural
material and of the methanolysis products. The original stereochemical
assignment of the alkaloids had been presumed to be one in which the
methine hydrogens in the pyrrolidine ring, and those adjacent to the
guanidine were all cis-, which was based on a previous literature
precedent.2 Subsequent work by Snider13 has shown that in the cases of batzelladine A
and D the original assignment was incorrect and the relative
stereochemistry of these metabolites was in fact trans. Both
batzelladine A and B contain two guanidinium ring systems connected by a
linear hydrocarbon chain, with the left-hand bicyclic guanidine being
similar in nature to crambescidin A 11. It is interesting to note
that the carbon skeleton present in the tricyclic guanidine nucleus of
batzelladine E, together with the alkene geometry is identical to that
found in ptilomycalin A 1 (Scheme
6).
 |
| | Scheme 6 | |
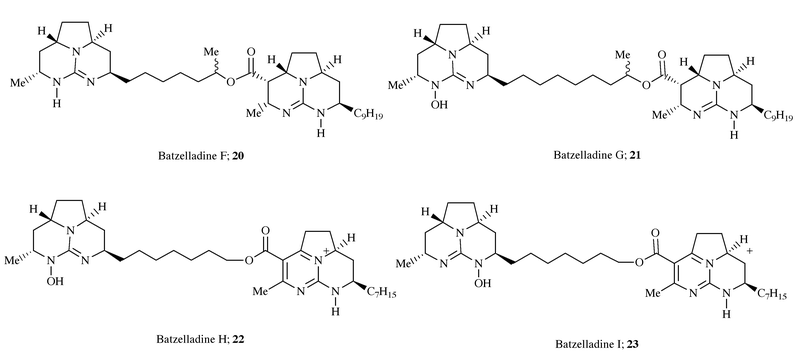
Following the isolation of these alkaloids, a further four batzelladine
metabolites, termed batzelladine F–I 20–23,
were obtained from the same source.14 These
were interesting in so much as they all contained two tricyclic guanidine
units one of which lacks an ester function, together with varying degrees
of unsaturation and the presence of N-hydroxylation in all but
20. The relative stereochemical assignment of these metabolites
was made by comparison with batzelladines A–E,4,13 however synthetic work15 has shown that the original assignment of the
left-hand tricycle of batzelladine F is possibly incorrect and that the
stereochemistry is in fact an all cis-arrangement of the methine
hydrogens (Scheme 7).
 |
| | Scheme 7 | |
As can be seen, these polycyclic guanidine alkaloids isolated from
marine sponges possess a wide range of biological activities as well as
intriguing molecular structures. It is thus not surprising that they have
attracted considerable interest from several synthetic research groups and
have also been the topic of some speculation
as to the exact biological role and mechanism of action of these
metabolites. The remainder of this review will focus on the synthetic
routes applied to these metabolites and will comment on work relating to
the biological aspects previously mentioned.
2 Synthetic contributions from the Snider
group
The Snider research group has made several significant contributions to
the synthesis of polycyclic guanidine alkaloids beginning with a synthesis,
in racemic form, of the metabolite ptilocaulin 2 and the
confirmation of its absolute stereo-chemistry by the synthesis of the
unnatural enantiomer 25via addition of guanidine to the
enone 2416 (Scheme 8).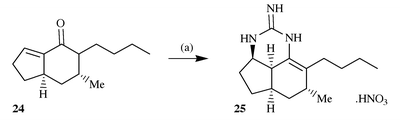 |
| | Scheme 8 Reagents and conditions: (a) Guanidine, PhH, reflux 25 h, then
HNO3 (1% aq); 40%. | |
A conceptually similar strategy has been adopted by Snider in his
synthetic approach to the crambescins, ptilomycalin A and the batzelladine
alkaloids. The first report in this area detailed the addition of
O-methylisourea to the enones 26 leading to the formation
of the dihydropyrimidines 27 after desilylation, which were then
converted into the guanidines 28 by treatment with methanolic
ammonia. These key intermediates were then converted into crambescins C1
13 and C2 14 by hydrogenolysis, crambescin A 11
by mesylation, cyclisation and hydrogenolysis and to crambescin B
12 by cyclisation under basic conditions and hydrogenolysis17 (Scheme
9).
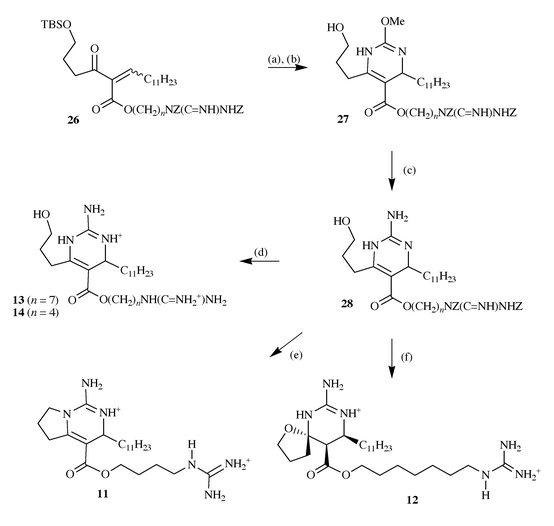 |
| | Scheme 9 Reagents and conditions: (a) 2 equiv.
O-methylisoureido sulfate, NaHCO3, DMF, 55 °C, 3 h,
94%; (b) HF, CH3CN, rt, 1.5 h, 88%; (c) NH3,
NH4OH, t-BuOH, MeOH, 60 °C, 3 days, (94% and 81%
loss of one of the two Z-groups is observed); (d) H2/Pd/C, HCl,
CHCl3 (88% and 91%); (e) MsCl, Et3N,
CH2Cl2, then Et3N, CHCl3,
Δ, 24 h, (78%) then H2/Pd/C, HCl, CHCl3, 3 h
(93%); (f) Et3N, Δ, CHCl3, 12 h, then
H2/Pd/C, HCl, CHCl3, rt, 3 h, (89% and 91%). | |
This work set the scene for a similar synthetic approach to ptilomycalin
A and preliminary model studies18 on the
bis-enone 31 prepared by Knoevenagel condensation of
β-ketoester 29 and aldehyde 30, illustrated that the
addition of O-methylisourea was a feasible process leading to the
bicyclic compound 32 as a 3∶1 mixture of trans- and
cis-isomers. This reaction appears to lead to the incorrect
stereochemistry at the pyrrolidine ring junction, however on reaction with
methanolic ammonia, which generates the guanidine functionality,
trans-32 undergoes isomerisation, presumably by an
elimination and readdition process, with concomitant cyclisation to form
the tricyclic model 33 of ptilomycalin A (Scheme 10).
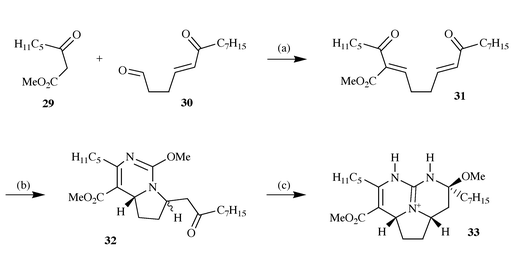 |
| | Scheme 10 Reagents and conditions: (a) Piperidine,
CH2Cl2, −20 °C, 2 days, 61%; (b) 2 equiv.
O-methylisoureido sulfate, NaHCO3, DMF, 50 °C, 2 h,
56%; (c) NH3, NH4OAc, MeOH, 60 °C, 4 days,
60%. | |
Following on from these preliminary studies Snider reported a synthesis
of the methyl ester of the pentacyclic nucleus of ptilomycalin A
38via a convergent and biomimetic 14-step route.19 The key steps of this route involved the
conversion of the bis-enone 34 to 38 in four steps. The
addition of O-methylisourea to 34 gave a 1∶4
mixture of the two cis diastereomers 35a and the two
trans diastereomers 35b in 52% yield. Ammonolysis
converted this mixture of isoureas into a 1:1 mixture of the two cis
diastereomeric guanidines 36a (cis,
β) and
36b (cis, α) in 72% yield. Deprotection of the
silyl ethers was accomplished by reaction with a 3:7 mixture of 50% aqueous
HF and acetonitrile. Treatment of the resulting crude mixture (containing
intermediate 37) with triethylamine in methanol gave a 60% yield
of an approximately 65% pure 1.3∶1 mixture of 38 and the
diastereomer 39 with an equatorial methyl ester. The
remaining material was thought to consist of tri- and tetracyclic compounds
from the undesired diastereomer 36b. Flash chromatography
succeeded in separating 38 and 39 but gave only
80–85% pure material. Purification of this mixture, therefore, was
best accomplished by treating the 1.3∶1 mixture of 38 and
39 with triethylamine in 1∶1 MeOH–H2O to
give tetracyclic alcohol 37. This was purified by flash
chromatography and recycled with triethylamine in methanol to again give a
1.3∶1 mixture of 38 and 39, which were separated to
give pure 38 (34% from 36a) and 39 (26% from
36a) (Scheme 11).
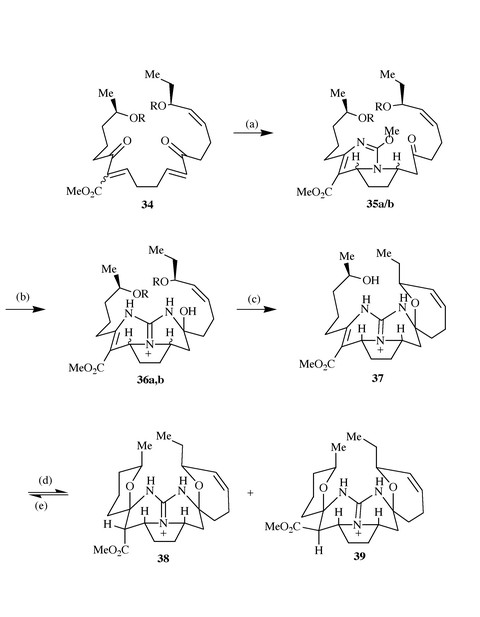 |
| | Scheme 11 Reagents and conditions: (a) O-methylisoureido
sulfate, i-Pr2EtN, DMSO, 80 °C, 1.5 h, 52%; (b)
NH3, NH4OAc, t-BuOH, 60 °C, 40 h, 72%;
(c) 3∶7 HF–CH3CN, −30 °C, 3 days; (d)
Et3N, MeOH, 60 °C, 20 h (78%); (e) Et3N,
MeOH–H2O (1∶1), 60 °C, 16 h, then (d),
38; 34% and 39; 26%. | |
Snider has also reported the first unequivocal synthesis (in racemic
form) of one of the batzelladine alkaloids, batzelladine E, and in doing so
was able to correct the structural misassignment of this metabolite which
was initially given an E-stereochemistry in the unsaturated side
chain.13b The synthesis begins
with the aldehyde 40 which was condensed with β-ketoester
41 under Knoevenagel conditions to give the enone 42.
Reaction of this enone with O-methylisourea followed by
ammonolysis gave the product 43 which on careful reduction led to
the tricyclic guanidine 44. Deprotection of the amine function was
followed by conversion to the terminal guanidine group, thus completing a
synthesis of batzelladine E in 9 steps and in 3% overall yield from a
commercially available starting material (Scheme
12).
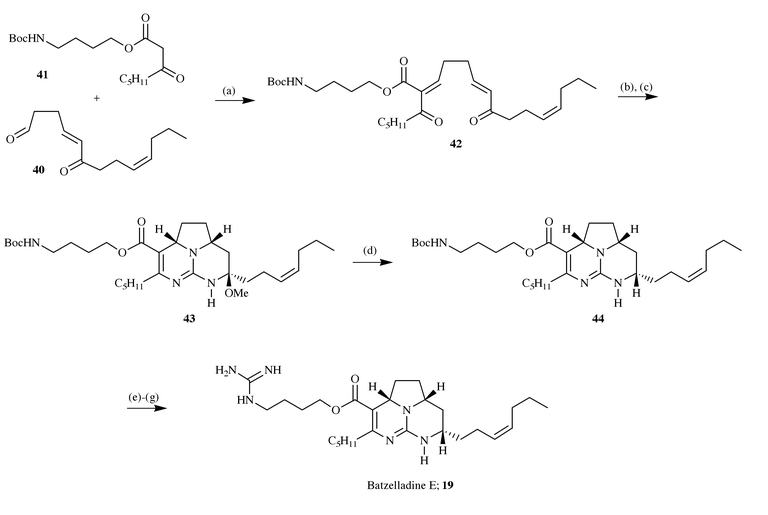 |
| | Scheme 12 Reagents and conditions: (a) 0.33 equiv. piperidine, 0.30
equiv. AcOH, CH2Cl2, −20 °C, 2 days; (b)
O-methylisourea, i-Pr2EtN, DMSO, 55 °C, 4
h; (c) NH3, NH4OAc, t-BuOH, 60 °C, 1 day
(14% from 40); (d) NaCNBH3,
NaH2PO4, MeOH, 25 °C, 88%; (e) 1:4
TFA–CH2Cl2, 25 °C, 5 min, 93%; (f)
(BocNH)2C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) S, 2-chloro-N-methylpyridinium
chloride, CH2Cl2, NEt3, 25 °C, 1 h,
64%; (g) 1∶1 TFA–CH2Cl2, 25 °C, 2 h,
90%. S, 2-chloro-N-methylpyridinium
chloride, CH2Cl2, NEt3, 25 °C, 1 h,
64%; (g) 1∶1 TFA–CH2Cl2, 25 °C, 2 h,
90%. | |
As mentioned previously, Snider had also reported13a the reassignment of the relative
stereochemistry of batzelladines A 15 and D 18. This was
accomplished by reaction of enone 45 with O-methylisourea
under standard conditions leading to a 35% yield of a 6∶1 mixture of
the dihydropyrimidine 46 and its cis-isomer. At this
point in the synthesis the ketone function was ‘protected’ by
reduction to the alcohol 47, which also serves to lock the
pyrrolidine in a cis-configuration, which was then converted to
the guanidine 48. Regeneration of the ketone function led to the
formation of the tricyclic guanidine 49 which was reduced and
saponified to give the carboxylic acid 50, a known degradation
product of batzelladines A and D, thus leading to the conclusion that the
stereochemistry in these metabolites was indeed trans- across the
pyrrolidine ring system (Scheme
13).
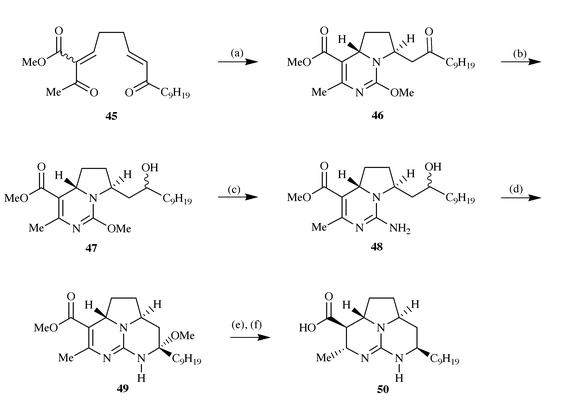 |
| | Scheme 13 Reagents and conditions: (a) O-methylisourea hydrogen
sulfate, i-Pr2EtN, DMSO, 75 °C, 5 h; (b)
NaBH4, i-PrOH, 25 °C; (c) NH3,
NH4OAc, MeOH, 60 °C, 2 days; (d) Dess–Martin,
CH2Cl2, 25 °C, then MeOH, 25 °C, 12 h; (e)
NaCNBH3, NaH2PO4, MeOH, 25 °C, 16 h
then 65 °C, 5 h; (f) NaOH, MeOH 25 °C, 18 h. | |
3 Synthetic contributions from the Overman
group
The Overman research group has also been actively pursuing the synthesis
of these polycyclic guanidine alkaloids, indeed, they have reported the
first total synthesis of ptilomycalin A 1 using a tethered
intramolecular Biginelli reaction.20,21 Preliminary studies on this reaction,20 which classically involves the condensation of
an aldehyde, a β-ketoester and a urea to give a
3,4-dihydro-1H-pyrimidin-2-one, had indicated its potential as a
synthetic methodology. A subsequent report21 illustrated that reaction of 51, which
represents the urea and masked aldehyde portions of the reaction, with the
β-ketoester 52 in the presence of morpholinium acetate
(typical Knoevenagel conditions), led to the formation of the bicyclic
pyrimidin-2-one 53. This product was cyclised to give the lower
half of the pentacyclic guanidine core 54 by treatment under
acidic conditions and converted to the aldehyde 55via
Swern oxidation and O-methylation with MeOTf. These two
intermediates differ from the corresponding portion in ptilomycalin A only
by the stereochemistry at the ester position. Introduction of the upper
half of the pentacycle was effected by reaction with the Grignard reagent
58 followed by Swern oxidation to the ketone 56; this was
followed by deprotection of the silyl protecting group and treatment with a
mixture of ammonia and ammonium acetate to give 57. Routine
modification of the ester function to a protected spermidine unit was
followed by epimerisation of the ester stereocentre by treatment with
triethylamine and finally deprotection, leading to the first total
synthesis of ptilomycalin A 1 (Scheme
14).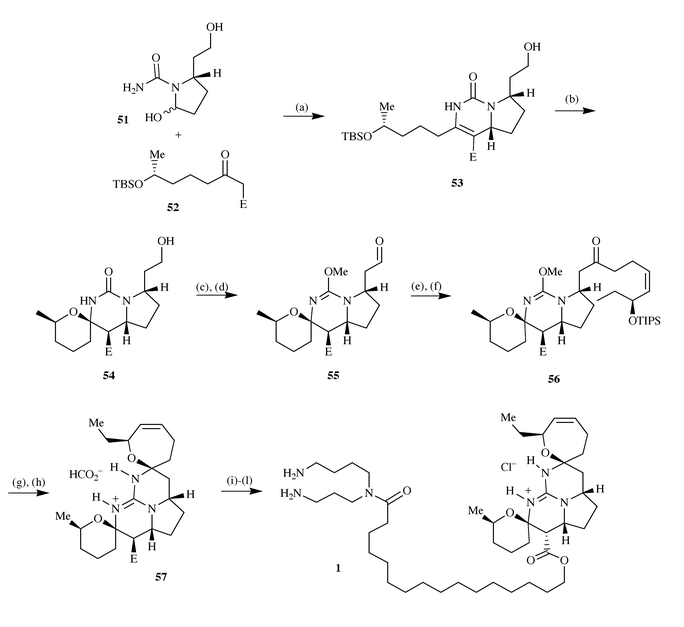 |
| | Scheme 14 Reagents and conditions: (a) Morpholine, AcOH, EtOH,
Na2SO4, 70 °C, 61%; (b) PPTS, MeOH, 50 °C,
then p-TosOH, CHCl3, 23 °C, 96%; (c) Swern
oxidation; (d) MeOTf, 2,6-di-tert-butylpyridine,
CH2Cl2, 23 °C, 67%; (e)
(S)-Z-EtCH(OTIPS)CHCHCH2CH2
MgBr (58), THF, −78 °C; (f) Swern oxidation, 58%
(2 steps); (g) TBAF; (h) NH3, NH4OAc,
t-BuOH, 60 °C, 51% (2 steps); (i)
Pd(Ph3P)4, pyrrolidine, MeCN, 23 °C, 75%; (j)
BocHN(CH2)3NH(CH2)4NHBoc, EDCl,
DMAP, CH2Cl2, 23 °C, 60%; (k) NEt3,
MeOH, 65 °C, 50%; (l) HCO2H, 23 °C, 100%. E =
CO2(CH2)15CO2Allyl. | |
Overman has also applied the tethered intramolecular Biginelli reaction
to the synthesis of tricyclic systems similar to those observed in the
batzelladines and has reported that reaction of bicyclic guanidine
59, prepared in 32% yield from nonan-2-one, with methyl
acetoacetate led to the formation of the tricycle 60 as a
10∶1 mixture of cis- and trans-isomers in 94%
yield.13c This structure is
identical to a reported4 degradation product
of batzelladine B in all respects, including the sign of rotation, thus
confirming the absolute stereochemistry of the batzelladines to be that
illustrated (Scheme 15).
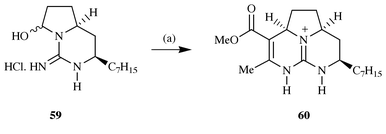 |
| | Scheme 15 Reagents and conditions: (a) Methyl acetoacetate, morpholinium
acetate (1 equiv.), CF3CH2OH,
Na2SO4, 90 °C, 36 h, 94%. | |
Interestingly Overman has also reported that by judicious choice of
reaction conditions it is possible to tune the selectivity of this reaction
to obtain either the cis- or trans-pyrrolidine
geometry.22 Thus treatment of either the
urea derivative 61 (X = O, R = H or Bn) with benzyl acetoacetate
under Knoevenagel conditions leads to the formation of
cis-62 and trans-62 (X = O, R = H or
Bn) in a 4∶1 ratio (80 and 81% yield respectively). However on
reaction of 61 (X = O, R = Bn) and benzyl acetoacetate with the
mild dehydrating agent polyphosphate ester (PPE) the stereoselectivity was
reversed leading to a 4∶1 ratio of products with the
trans-isomer predominating. Interestingly, treatment of the
guanidine 61 (X = NH·HCl, R = OH) under identical
Knoevenagel conditions led to the formation of trans- 62
(X = NH·HCl, R = OH) exclusively in 42% yield, which is in complete
contrast to the stereoselection observed with ureas. Further work with the
N-sulfonylguanidine 61 (X = MtrN, R = H, Bn) led to the
formation of cis-62 and trans-62 in a
6–7∶1 ratio (X = MtrN, R = H, Bn; 61 and 84% yield
respectively) under Knoevenagel conditions, but again reaction of
61 (X = MtrN, R = Bn) with PPE led to a reversal of selectivity
with the trans-62 (X = MtrN, R = Bn) predominating in a
20∶1 ratio (61% yield) (Scheme
16).
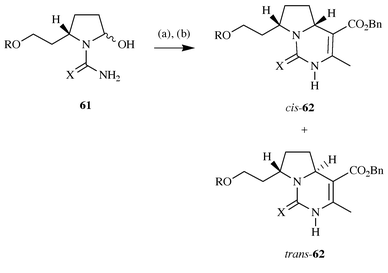 |
| | Scheme 16 Reagents and conditions: (a) Benzyl acetoacetate (1.5 equiv.),
morpholinium acetate (1.5 equiv), CF3CH2OH,
Na2SO4, 60 °C, 48 h; (b) PPE,
CH2Cl2, 23 °C, 48 h. | |
4 Synthetic contributions from the Murphy
group
Within our own research group we have been pursuing the synthesis of
both ptilomycalin A and the batzelladine alkaloids utilising a strategy
based upon the double 1,4-addition of guanidine to bis-enones. Preliminary
studies23 illustrated that the formation of
the pentacyclic guanidines 66 and 69 was possible using
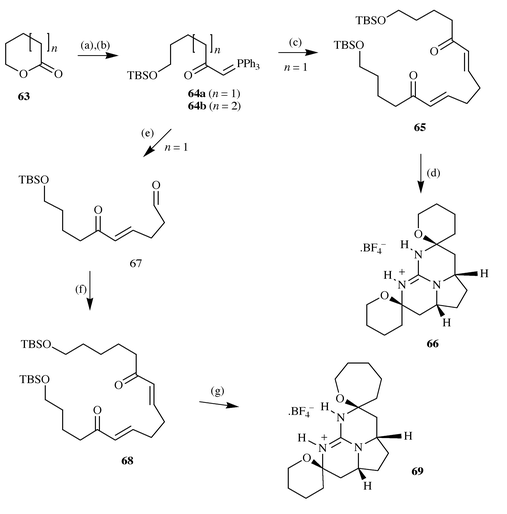
this strategy. Synthesis of the substrate for these model studies was
straightforward in that reaction of lactones 63 with two
equivalents of methylenetriphenylphosphorane followed by silyl protection
of the intermediate phosphonium alkoxide gave the phosphoranes 64.
Wittig reaction of 64a with 0.4 equivalents of succinaldehyde gave
the symmetrical bis-enone 65 in 54% yield. Further reaction of
this with one equivalent of guanidine, followed by removal of solvent,
deprotection/cyclisation with methanolic HCl and counter ion exchange,
afforded two products identified as the cis-product 66
and the corresponding trans-pentacycle in an approximate 4∶1
ratio, from which the major product could be isolated in 25% yield by
recrystallisation.Similarly reaction of the phosphorane 64a with an excess of
succinaldehyde led to the formation of the aldehyde 67 in 43%
yield from the lactone starting material; reaction of this with phosphorane
64b gave the unsymmetrical bis-enone 68. Reaction of
68 with guanidine under identical conditions to those previously
employed, led to the formation of two pentacyclic guanidines in an
approximate 4∶1 ratio and from which the major isomer 69
could be obtained by crystallisation in 20% yield (Scheme 17).
 |
| | Scheme 17 Reagents and conditions: (a) 2 equiv.
CH2PPh3, THF, −78 °C; (b) TBDMSCl,
imidazole, DMF; (c) 0.4 equiv. succinaldehyde, THF, 48 h; 54% overall; (d)
(i) guanidine, DMF, 3 h, (ii) MeOH, HCl, 0 °C–rt, 24 h; (iii) aq.
NaBF4 (sat.), (iv) trituration and crystallisation; 25% overall;
(e) steps (a), (b) then 10 equiv. succinaldehyde, THF, 43%; (f)
64b, THF, 48 h; 37%; (g) as (d) 20%. | |
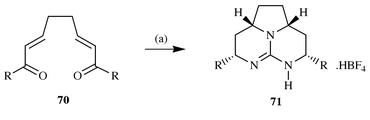
Following on from this work we also reported that the addition of
guanidine to a series of bis-enones 70 followed by reduction with
sodium borohydride led to the formation of the tricyclic guanidines
71, which, before the structural reassignments reported by
Snider,13 were thought to be models of the
saturated batzelladine alkaloids A and D24
(Scheme 18).
 |
| | Scheme 18 Reagents and conditions: (a) (i) Guanidine, DMF, 0
°C–rt, 5–8 h, (ii) 3∶1∶3
DMF–H2O–MeOH, then NaBH4, 16 h, (iii) HCl
(aq), (iv) aq. NaBF4 (sat.). R = Me,
C5H11, C9H19, Ph, (22–33%
yield). | |
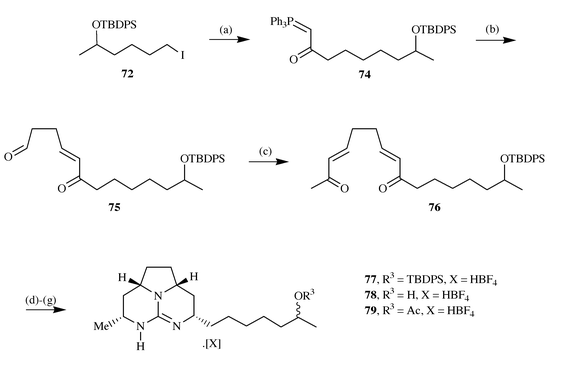
With the isolation of batzelladine F, it became apparent that there was
a strong correlation in structure between the left-hand portion of this
molecule and the model compounds previously prepared by us;15a we thus embarked upon a synthesis of
the left-hand sub unit of this metabolite employing this
methodology.15b Reaction of the
iodide 72 with the anion generated from deprotonation of
phosphorane 73 led to the phosphorane 74, which on
reaction with excess succinaldehyde gave the aldehyde 75.
Subsequent reaction of this with a further equivalent of phosphorane
73 led to the formation of bis-enone 76, which on
reaction with guanidine under standard conditions gave the tricycle
77. Comparison of the spectral data of 77 or the
corresponding deprotected analogue 78 or acetate 79,
illustrated a strong correlation between these materials and the naturally
occurring batzelladine F thus leading to the conclusion that the reported
stereochemistry is incorrect and is actually as illustrated25 (Scheme
19).
 |
| | Scheme 19 Reagents and conditions: (a) CH3COCHPPh3
(73), n-BuLi, THF, −78 °C–rt; (b)
succinaldehyde, CH2Cl2, 24 h, 54% for 2 steps; (c)
73, CH2Cl2, 24 h, 91%; (d) (i) guanidine,
DMF, 0 °C, 5 h; (ii) 3∶1∶3
DMF–H2O–MeOH, then NaBH4, 16 h; (iii) HCl
(aq), (iv) aq. NaBF4 (sat); 77, 29% overall; (e) (i)
MeOH–HCl, (ii) aq. NaBF4 (sat.), 78, 91%; (f)
Ac2O–Py, then HCl (2 M), 41%; (g) aq. NaBF4
(sat), 79,100%. | |
5 Synthetic contributions from the Hiemstra
group
A recent publication from the Hiemstra group describes the preparation
of tricyclic guanidines from substituted pyrrolidin-2-ones utilising an
N-acyliminium ion coupling reaction with silyl enol ethers and a
direct guanylation with bis-Boc-thiourea and mercury(II)
chloride.26 They reported that reaction of
the silyl enol ether 80 with lactam 81 led to the
formation of the substituted lactam 82 in 63% yield. After
conversion of 82 into the corresponding thiolactam, an Eschenmoser
sulfide contraction procedure led to the formation of the vinylogous amide
83. Reduction and N-Boc protection of 83 gave
the substrate 84 which was subjected to a three stage procedure
which included protected guanidine 85 as an intermediate. This was
cyclised under acidic conditions to give the tricyclic guanidine
86 in 33% overall yield, together with several other
trans-substituted guanidines which could be recycled to give
further 86 by treatment with ammonia and ammonium acetate in
methanol at 60 °C26 (Scheme 20). |
| | Scheme 20 Reagents and conditions: (a) TMSOTf, −78 °C–rt,
CH2Cl2, 18 h, 63%; (b) Lawesson’s reagent,
PhCH3, 80 °C, 10 min, 91%; (c) PhCOCH2Br,
Et2O, rt, 18 h; (d) Et3N,
CH2Cl2, rt, 2 h, 83%; (e) PPh3,
CHCl3, 60 °C, 18 h, 82%; (f) NaBH3CN, 3∶1
AcOH–THF, 0 °C, 40 min, 99%; (g) Boc2O, DIPEA, THF,
rt, 18 h, 91%; (h) PCC, CH2Cl2, mol. sieves (4
Å), rt, 3 h, 91%; (i) CH(OMe)3,
H2SO4 (cat), MeOH, 50 °C, 5 h; (j)
SC(NHBoc)2, HgCl2, Et3N, DMF, 0
°C–rt, 18 h; X = O, (OMe)2; (k) HCl, MeOH, rt, 3 h,
33% for three steps. | |
This synthesis is obviously applicable to the preparation of both
ptilomycalin A 1 and the batzelladine alkaloids. Indeed Hiemstra
has further reported27 that
bis-acetoxylactam 87 reacts with the silyl enol ether 88
in a stereocontrolled fashion to give 89, which is a potential
precursor of 1 (Scheme
21).
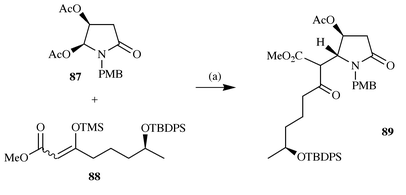 |
| | Scheme 21 Reagents and conditions: (a) TMSOTf, −78 °C–rt,
CH2Cl2, DIPEA, 1 h, 75%. | |
6 Synthetic contributions from the Rama Rao
group
The Rama Rao research group has also reported28 an enantiospecific synthesis of the tricyclic
guanidine portion of batzelladine A, which was based upon the original
stereochemical assignment made by Patil,4
and thus targeted an all cis-arrangement of hydrogens in the final
product. In common with Hiemstra, the synthesis proceeds via a
lactam, 91 prepared in eight steps from the azetidinone derivative
90, and introduction of the side chain was effected using an
Eschenmoser sulfide contraction reaction, leading to the
α,β-unsaturated ketone 92. After reduction of the
alkene and ketone functions and N-Boc protection of the
pyrrolidine nitrogen, the introduction of the remaining nitrogen containing
groups was accomplished by sequential nucleophilic displacement of the
secondary alcohols present on each side chain with azide (under Mitsunobu
conditions), reduction and N-boc protection, ultimately leading to
azide 93. This was converted to the cyclic urea 94 using
1,1′-carbonyldiimidazole, which in turn was cyclised by treatment
with dimethylsulfate and hydrogenation to give the guanidine 95,
after removal of the silyl protecting group (Scheme 22).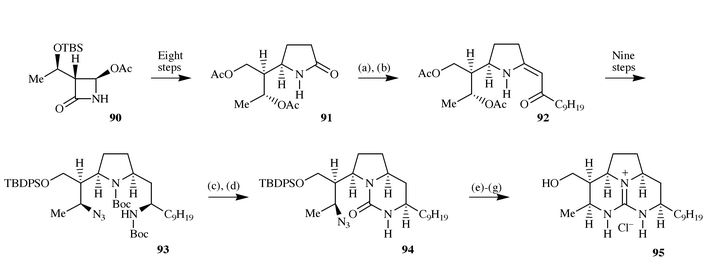 |
| | Scheme 22 Reagents and conditions: (a)
C9H19COCH2Br,
CH2Cl2, rt, 30 min KHCO3; (b)
PPh3, t-BuOK, t-BuOH,
C6H6, heat, 65%; (c) TFA,
CH2Cl2, 0 °C–rt, 30 min; (d)
(imid)2CO, THF, 0 °C–rt, 65%; (e)
Me2SO4, C6H6, heat, 16 h; (f)
H2, Pd/BaSO4, MeOH, 12 h, 65%; (g) HCl (1 M), MeOH,
50 °C, 2 h, 90%. | |
7 Ptilomycalin A as a potential host
molecule
One important feature of the chemistry of guanidinium compounds is their
ability to interact with anionic species, indeed, this is a key feature in
the function of the amino acid arginine whose guanidine group serves as a
key binding site for carboxylate and phosphate containing substrates in a
wide range of biological applications.29 It
is apparent that ptilomycalin A 1 has an extraordinary molecular
architecture in which the pentacyclic core (Fig.
1) can be considered to have a ‘cage’ like structure
capable of binding strongly to an anion and possibly encapsulating it,
indeed, Kashman referred to this core as the ‘vessel’ part of
the molecule linked via the aliphatic ‘chain’ to a
spermidine ‘anchor’.2b
In fact, one interesting feature of this metabolite is its structural
similarity to abiotic guanidine based anionic receptor molecules.30 Based on this hypothesis, Kashman2b investigated the complexing ability of a
trifluoroacetate derivative of 1 with various organic carboxylates
and determined a scale of binding ability for N-acetylamino acids
which was estimated to be as follows:
L-N-acetylmethionate ≈
L-N-acetylvalinate >
L-N-acetylalanate ≈
L-N-acetylisoleucinate >>
L-N-acetylglycinate. In addition, there is evidence
suggesting that the spermidine unit may also be involved in the binding of
anionic species to ptilomycalin A.31 It is
also interesting to note that ptilomycalin A, despite containing several
polar functional groups, is a relatively non-polar molecule which is freely
soluble in organic solvents such as chloroform.2a These properties suggest an anionic
binding capability possibly linked to strong lipophillic behaviour.21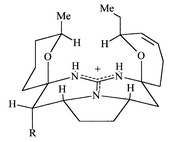 |
| | Fig. 1 | |
It has been speculated32 that the
enclosed ionic ‘cage’ at the central guanidine sub-unit might
indeed be acting as a specific anion recognition site thereby conferring
much of the biological activity found in these compounds. In relation to
this, it is interesting to note that the subsequently isolated
13,14,15-isocrambescidin 800 10 is substantially less cytotoxic to
L1210 cells than other crambescidins and has no observed antiviral activity
against HSV-1;10 this reduced activity may
be due to the lack of this structural feature (Fig.
2).
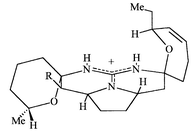 |
| | Fig. 2 | |
We have reported32 an interesting
behaviour pattern in the interaction between the guanidinium and
fluoroborate ions in the model compounds 96, 66 and
97. Fluoroborate can undergo a similar interaction with a
guanidinium ion to that of the bidentate ligating interaction that is
observed with a carboxylate or a phosphate. It was thus surprising to find
that in compound 96 only one of the fluorine atoms of the
fluoroborate anion was involved in a strong hydrogen bonding interaction
with the guanidinium cation (to both N–H bonds). This observation led
us to suppose that the guanidinium cavity of 96 was not of
sufficient size to accommodate the fluoroborate anion. This behaviour was
however not observed in the pentacyclic 6,6,5,6,6 model compound
66 which corresponds more closely to the structure of ptilomycalin
A. In this case, the fluoroborate anion was involved in two separate
non-symmetrical hydrogen bonding interactions, but was unable to achieve
co-planarity with the guanidinium ion. However, near co-planarity was
achieved in the case of the pentacyclic 7,6,5,6,7 model 97 which
undergoes an identical hydrogen bonding pattern to 66 (Scheme 23). These observations offer some support
to the theory of the ‘cage’ portion of ptilomycalin A being
involved in a recognition process and that the structure of 1
represents an optimum host design for an as yet undetermined guest
molecule.
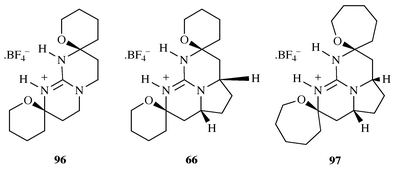 |
| | Scheme 23 | |
A further report from Hart and Grillot33
describes the synthesis of an analogue 98 (Scheme 24), of ptilomycalin A 1, which
models the guanidine core, chain and spermidine sub-units of the natural
material and was prepared in a 12 step convergent sequence from acyclic
precursors via a cyclic thiourea derivative. This molecule was
prepared in an attempt to mimic the biological activity found in the
natural system, however, it was found to be unstable and underwent an
unidentified decomposition process over a period of a few weeks which
precluded biological evaluation. It was suggested that this process
involved cleavage of the ester linkage and the authors speculated that the
role of the spiro-N,O-acetals present in ptilomycalin A 1
might be to protect the ester linkages from hydrolysis or
aminolysis.
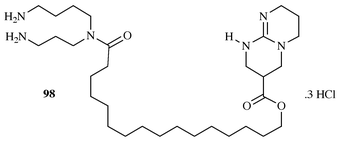 |
| | Scheme 24 | |
8 Conclusion
Ptilomycalin A and related molecules are obviously of considerable
interest from both a synthetic and biological perspective. The current
state of the synthetic work offers several strategies for the synthesis of
these metabolites and analogues thereof, with several total syntheses
having been reported. The biological activities of these metabolites ranges
over a broad spectrum, however, little is known about the exact mechanism
of these activities despite much speculation. We believe that in the near
future this area of investigation, together with continued synthetic
efforts, will offer a valuable insight into these intriguing
metabolites.References
-
For leading references see: R. G. S. Berlinck, Nat. Prod. Rep.,
1995, 377. Search PubMed.
-
(a) Y. Kashman, S. Hirsh, O. J. McConnell, I. Ohtani, T. Kusumi and H. J. Kakisawa, J. Am. Chem. Soc., 1989, 111, 8925;;
(b) I. Ohtani, T. Kusumi, H. Kakisawa, Y. Kashman and S. Hirsh, J. Am. Chem. Soc., 1992, 114, 8472 CrossRef CAS.
- R. G. S. Berlinck, J. C. Braekman, D. Daloze, K. Hallenga, R. Ottinger, I. Bruno and R. Riccio, Tetrahedron Lett., 1990, 31, 6531 CrossRef CAS.
- A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. De Brosse, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Potts, J. Org. Chem., 1995, 60, 1182 CrossRef CAS.
- E. Palagiano, S. De Marino, L. Minale, R. Riccio, F. Zollo, M. Iorizzi, J. B. Carré, C. Debitus, L. Lucarain and J. Provost, Tetrahedron, 1995, 51, 3675 CrossRef CAS.
- G. C. Harbour, A. A. Tymiak, K. L. Rinehart, P. D. Shaw, R. G. Hughes, S. A. Mizsak, J. H. Coats, G. E. Zurenko, L. H. Li and S. L. Kuentzel, J. Am. Chem. Soc., 1981, 103, 5604 CrossRef CAS.
- E. A. Jares-Erijman, R. Sakai and K. L. Rinehart, J. Org. Chem, 1991, 56, 5712 CrossRef CAS.
- R. Tavares, D. Daloze, J. C. Braekman, E. Hadju, G. Muricy and R. W. M. van Soest, Biochem. Syst. Ecol., 1994, 22, 645 CrossRef CAS.
- R. G. S. Berlinck, J. C. Braekman, D. Daloze, I. Bruno, R. Riccio, S. Ferri, S. Spampinato and E. Speroni, J. Nat. Prod., 1993, 56, 1007 CrossRef CAS.
- E. A. Jares-Erijman, A. L. Ingrum, J. R. Carney, K. L. Rinehart and R. Sakai, J. Org. Chem., 1993, 58, 4805 CrossRef CAS.
-
(a) R. G. S. Berlinck, J. C. Braekman, D. Daloze, I. Bruno, R. Riccio, D. Rogeau and P. Amade, J. Nat. Prod., 1992, 55, 528;;
(b) E. A. Jares-Erijman, A. A. Ingrum, F. Sun and K. L. Rinehart, J. Nat. Prod., 1993, 56, 2186 CrossRef CAS.
- Some confusion arose in the identification of the sponge
Batzella sp.; a comparison of a specimen with a known specimen of
Ptilocaulis sp. illustrated that the two were in fact the
same.4.
-
(a) B. B. Snider, J. Chen, A. D. Patil and A. J. Freyer, Tetrahedron Lett., 1996, 37, 6977 CrossRef CAS;
(b)
the alkene stereochemistry in batzelladine E has also been recently
reassigned, see, B. B. Snider and J. Chen, Tetrahedron Lett.,
1998, 39, 5697; Search PubMed;
(c)
the absolute stereochemistry of batzelladine B has been determined by
synthesis of a degradation product and is as shown in the scheme, however
this is the opposite of the arbitrary stereochemistry chosen by the authors
of reference 4; A. S. Franklin, S. K. Ly, G. H. Mackin, L. E. Overman and A. J. Shaka, J. Org. Chem.,
1999, 64, 1512. Search PubMed.
- A. D. Patil, A. J. Freyer, P. B. Taylor, B. Carté, G. Zuber, R. K. Johnson and D. J. Faulkner, J. Org. Chem, 1997, 62, 1814 CrossRef CAS.
-
(a) G. P. Black, P. J. Murphy and N. D. A. Walshe, Tetrahedron, 1998, 54, 9481; CrossRef CAS;
(b) G. P. Black, P. J. Murphy, A. Thornhill, N. D. A. Walshe and C. Zanetti, Tetrahedron, 1999, 55, 6547 CrossRef CAS.
- B. B. Snider and W. C. Faith, J. Am. Chem. Soc., 1984, 106, 1443 CrossRef CAS.
-
(a) B. B. Snider and Z. Shi, J. Org. Chem., 1992, 57, 2526;;
(b) B. B. Snider and Z. Shi, J. Org. Chem., 1993, 58, 3828 CrossRef CAS.
- B. B. Snider and Z. Shi, Tetrahedron Lett., 1993, 34, 2099 CrossRef CAS.
- B. B. Snider and Z. Shi, J. Am. Chem. Soc., 1994, 116, 549 CrossRef CAS.
- L. E. Overman and M. H. Rabinowitz, J. Org. Chem., 1993, 58, 3235 CrossRef CAS.
- L. E. Overman, M. H. Rabinowitz and P. A. Renhowe, J. Am. Chem. Soc., 1995, 117, 2657 CrossRef CAS.
- A. I. McDonald and L. E. Overman, J. Org. Chem., 1999, 64, 1520 CrossRef CAS.
-
(a) P. J. Murphy, H. L. Williams, M. B. Hursthouse and K. M. A. Malik, J. Chem. Soc., Chem. Commun., 1994, 119; Search PubMed;
(b) P. J. Murphy and H. L. Williams, J. Chem. Soc., Chem. Commun., 1994, 819; RSC;
(c) P. J. Murphy, H. L. Williams, D. E. Hibbs, M. B. Hursthouse and K. M. A. Malik, Tetrahedron, 1996, 52, 8315 CrossRef CAS.
- G. P. Black, P. J. Murphy, N. D. A. Walshe, D. E. Hibbs, M. B. Hursthouse and K. M. A. Malik, Tetrahedron Lett., 1996, 37, 6943 CrossRef CAS.
- Comments of this nature are also made in reference 8 of the recent
paper by Overman.13c.
- S. Louwrier, M. Ostendorf, A. Tuynman and H. Hiemstra, Tetrahedron Lett., 1996, 37, 905 CrossRef CAS.
-
(a) S. Louwrier, M. Ostendorf, A. Boom, H. Hiemstra and W. N. Speckamp, Tetrahedron, 1996, 52, 2603; CrossRef;
(b) S. Louwrier, A. Tuynman and H. Hiemstra, Tetrahedron, 1996, 52, 2629 CrossRef CAS.
- A. V. Rama Rao, M. K. Gurjar and J. Vasudevan, J. Chem. Soc., Chem. Commun., 1995, 1369 RSC.
- C. L. Hannon and E. V. Anslyn, Bioorg. Chem. Front., 1993, 3, 193 Search PubMed.
- For leading references see: F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS.
- I. Ohtani, T. Kusumi and H. Kakisawa, Tetrahedron Let., 1992, 33, 2525 CrossRef CAS.
- P. J. Murphy, H. L. Williams, D. E. Hibbs, M. B. Hursthouse and K. M. A. Malik, Chem. Commun., 1996, 445 RSC.
-
(a) A.-L. Grillot and D. J. Hart, Tetrahedron, 1995, 51, 11377; CrossRef CAS;
(b) A.-L. Grillot and D. J. Hart, Heterocycles, 1994, 39, 435 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.