Coordination of carboxamido nitrogen to tervalent iron: insight into a new chapter of iron chemistry
Dana S. Marlin and Pradip K. Mascharak*
Department of Chemistry and Biochemistry, University of California, Santa Cruz, California 95064, USA
First published on UnassignedUnassigned11th January 2000
Abstract
Although coordination of carboxamido nitrogen to Fe(III) center has been assumed to be improbable, research work during the past few years has demonstrated that Fe(III) complexes with ligated carboxamido nitrogens can be readily synthesized. The Fe(III)–Namido bond distances lie in the range of 1.8–2.2 Å in the various low spin and high spin Fe(III) complexes. These complexes are stable in aqueous media and their redox parameters indicate that the carboxamido nitrogens provide significant stability to the Fe(III) center.
 Dana S. Marlin Dana S. Marlin | Dana Marlin was born in Newport Beach, California in 1972 and raised in Malta. He received his BS in Biochemistry from the California Polytechnique University at San Luis Obispo in 1996. Since then, he has been working as a graduate student in the research group of Professor Mascharak at the University of California, Santa Cruz. His research interests include syntheses and characterization of iron complexes that exhibit reactivity toward small molecules like dioxygen and NO. His hobbies include windsurfing and sailing. |
 Pradip K. Mascharak Pradip K. Mascharak | Pradip Mascharak was born in Jaipur, India in 1953. He received his PhD from the Indian Institute of Technology, Kanpur in 1979. In the same year, he joined the research group of Professor Richard Holm at Stanford University and later moved to Harvard University. He also worked with Professor Steve Lippard at the Massachusetts Institute of Technology for two years before joining the University of California, Santa Cruz in late 1984. He is currently a Professor of Chemistry and Biochemistry at UCSC. Modeling of active sites of metalloenzymes, design of antitumor drugs and catalysts for hydrocarbon oxidation are the major focus of his research. His hobbies include photography and gardening. |
1 Introduction
In the excellent review by Sigel and Martin on the chemistry of metal–peptide complexes,1 a problem with respect to the complexation of Fe(II) and Fe(III) by peptido nitrogen was discussed. Since the pH necessary for deprotonation of the peptide nitrogen is too high for Fe(II) and Fe(III) to exist in aqueous solution (both ions precipitate as hydroxides at pH![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 3),2 it had generally been concluded that ligation of
peptido nitrogen to iron was improbable. However, our research work in the
area of iron bleomycin and related chemistry revealed that small ligands
with peptide NH groups readily coordinate to both Fe(II) and
Fe(III) centers in their deprotonated forms.3–8 These findings along with
reports from other groups9–15 have provided some insight into the nature of
the iron–peptide bond. In addition, the recent identification of
coordination of peptido nitrogen from the peptide backbone to iron centers
in the p-cluster of nitrogenase16 and in
the mononuclear non-heme iron center of nitrile hydratase17,18 has raised more interest in this area.
During the past few years, several complexes of tervalent iron with
carboxamido nitrogen (peptido nitrogen is a subset of carboxamido nitrogen)
donors have been structurally characterized. The majority of these
complexes have two carboxamido nitrogens3–6,8–10 and the rest have four such
donors in the first coordination sphere.7,11–14 In contrast, Fe(III)
complexes with an odd number of coordinated carboxamido nitrogens have been
limited19,20 and no structural
information is available so far. Collectively, the spectral and structural
data for the Fe(III) complexes now indicate that coordination of
carboxamido nitrogens to an Fe(III) center is not
unusual and the coordination chemistry of Fe(III) complexes
with peptides could be developed much like any other ligand. The purpose of
this review is to highlight the synthetic routes to such complexes and to
determine the effect(s) of the coordinated carboxamido group on their
stability and redox properties.
3),2 it had generally been concluded that ligation of
peptido nitrogen to iron was improbable. However, our research work in the
area of iron bleomycin and related chemistry revealed that small ligands
with peptide NH groups readily coordinate to both Fe(II) and
Fe(III) centers in their deprotonated forms.3–8 These findings along with
reports from other groups9–15 have provided some insight into the nature of
the iron–peptide bond. In addition, the recent identification of
coordination of peptido nitrogen from the peptide backbone to iron centers
in the p-cluster of nitrogenase16 and in
the mononuclear non-heme iron center of nitrile hydratase17,18 has raised more interest in this area.
During the past few years, several complexes of tervalent iron with
carboxamido nitrogen (peptido nitrogen is a subset of carboxamido nitrogen)
donors have been structurally characterized. The majority of these
complexes have two carboxamido nitrogens3–6,8–10 and the rest have four such
donors in the first coordination sphere.7,11–14 In contrast, Fe(III)
complexes with an odd number of coordinated carboxamido nitrogens have been
limited19,20 and no structural
information is available so far. Collectively, the spectral and structural
data for the Fe(III) complexes now indicate that coordination of
carboxamido nitrogens to an Fe(III) center is not
unusual and the coordination chemistry of Fe(III) complexes
with peptides could be developed much like any other ligand. The purpose of
this review is to highlight the synthetic routes to such complexes and to
determine the effect(s) of the coordinated carboxamido group on their
stability and redox properties.2 Synthetic methods
To date, the ligands that have been employed to isolate Fe(III) complexes are cyclic or acyclic polydentate ligands with one, two, or four carboxamide groups.† Since the focus of this review is on Fe(III) complexes for which the structures are known, the list of such ligands is limited to the ones shown in Fig. 1. These ligands were designed for various reasons in different laboratories. For example, the tridentate ligands PypepH and PrpepH were designed to establish the mode of binding of bleomycin to iron3,4 while bpcH2 was synthesized as part of studies on oxidation of hydrocarbons by metal complexes.9,10 Along the same line, the S-containing ligands PypepSH25 and PypepS2H46 were specially prepared to model the active site of the enzyme nitrile hydratase. The ligands Py3PH2, MePy3PH2,7 LH2,11 PypepOH2,21 and POPYH48 were however intended for the development of the coordination chemistry of Fe(III) complexes with carboxamido nitrogens as donors. And finally, the macrocyclic tetraamido ligands H4[MAC*],14 and H4[3]12,13 were developed by Collins and coworkers to stabilize metal ions in high oxidation states.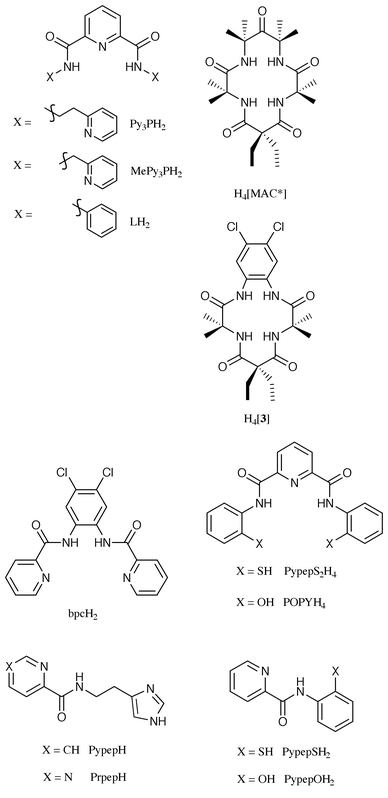 | ||
| Fig. 1 Ligands used in the syntheses of Fe(III) complexes. | ||
Although the synthetic strategies for successful isolation of Fe(III) complexes with ligated carboxamido nitrogens have just begun to emerge, some common themes in their syntheses have already been recognized. The known Fe(III) complexes have been synthesized by either of the two following methods. The first one involves initial formation of the Fe(II) species under anaerobic conditions followed by oxidation to the Fe(III) complex. The second approach involves the synthesis of the Fe(III) complex directly from the reaction of a suitable Fe(III) salt with the deprotonated ligand. The correct choice of base and solvent, as well as the appropriate Fe(III) salt are the crucial factors for success with this method.
The choice of the Fe(III) source depends on the type of complex one decides to synthesize. For the bis complexes with two carboxamido nitrogens around Fe(III), FeCl33 or [Et4N][FeCl4]4–6 is preferred. However, attempts to prepare the bis complexes with four carboxamido nitrogens involving the ligands Py3PH2 and MePy3PH2 invariably failed.7 We have recently discovered that [Fe(DMF)6](ClO4)3 is a very convenient starting material for the syntheses of Fe(III) complexes with these multidentate ligands7 and others like PyPepS2H4 and POPYH4.6,8 This Fe(III) starting material can be readily prepared and it is indefinitely stable and not very hygroscopic. One may also isolate the Fe(III) complex via oxidation of the corresponding Fe(II) species. The tetracarboxamido macrocyclic complexes [Fe(3)(H2O)]− and [Fe(η4-MAC*)(Cl)]2− have been prepared from FeCl2 followed by air oxidation.12–14 [Fe(CH3CN)4](ClO4)2 is another Fe(II) starting material that has also been used in several cases.9,11
Both protic and aprotic solvents have been used in the syntheses of Fe(III) complexes with ligated carboxamido nitrogens. In our earlier work, protic solvents such as ethanol and methanol have been employed in the preparation of [Fe(Pypep)2]Cl and [Fe(Prpep)2]Cl.3,4 Che and coworkers have also used methanol in their synthesis of trans-[Fe(bpc)(1-MeIm)2]ClO4.10 For the remaining complexes, the two aprotic solvents N,N′-dimethylformamide (DMF) and acetonitrile have been used. The need for these aprotic media arises from the strongly basic conditions necessary to deprotonate the carboxamide nitrogen.1 Once the Fe(III)–Namido bonds are formed, the complexes are often indefinitely stable in various protic solvents including water. Indeed, some of the reported complexes have been manipulated further in water. For example, [Fe(3)H2O]− has been prepared from the [Fe(3)Cl]2− precursor via removal of the chloride anion with Ag+ in water.12,13 Also, the seven coordinate complexes Na[Fe(POPY)(1-MeIm)2] and Na3[Fe(POPY)(NCS)2] are converted to the corresponding aquo species in water. This transformation is reversible since the original complexes are recovered from such solutions upon addition of excess 1-MeIm or NaSCN.8 It is therefore evident that the inherent basicity of the carboxamido nitrogen is lowered considerably upon coordination to the Fe(III) center, a fact that prevents hydrolysis of these complexes in water.7
The choice of base is very crucial in all the syntheses mentioned above. The base must be sufficiently strong to deprotonate the peptide nitrogens, but must not react with the solvent. In protic solvents, one generally adds the metal source to the ligand prior to the addition of the base. It appears that initial coordination of the ligand to the Fe(III) center assists deprotonation of the carboxamide group. In such cases, amines like triethylamine or 1,8-bis(dimethylamino)naphthalene are good bases.3,4 In acetonitrile, other bases like CH3COONa have also been used.10 When the complexation reaction is performed in DMF, NaH is the base of choice. In such reactions, the base must be added to the ligand prior to the addition of the metal salt. The combination of NaH and DMF (or acetonitrile) is particularly favorable since coordination of solvent to the Na+ ions in the crystal lattice often helps crystallization of the Na+ salts of the anionic complexes. The strong base tert-butyllithium has been used to deprotonate the macrocyclic ligands H4[3]12,13 and H4[MAC*].14
Finally it is important to mention the need for anaerobic conditions while preparing the thiolato complexes [Fe(PypepS)2]− and [Fe(PypepS2)]−. Upon subsequent exposure to oxygen, these complexes are converted to the sulfinato derivatives [Fe(PypepSO2)2]− and [Fe(PypepS2O4)]−.6,22 Fe(III) complexes with no thiolato sulfur(s) in the first coordination sphere are however synthesized under aerobic conditions and the complexes are stable in air.
3 Structures
Of the twelve ligands listed in Fig. 1, the majority form six coordinate octahedral complexes with Fe(III) although some form complexes with coordination numbers of five and seven. In addition, the complexes that have been structurally characterized so far have the Fe(III) center coordinated to either two or four carboxamido nitrogen donors. For the sake of comparison, the structural features of these Fe(III) complexes with two carboxamido nitrogens and four carboxamido nitrogens are treated separately in the following sections. The average Fe(III)–Namido bond distances for these complexes are listed in Table 1. As expected, the average Fe(III)–Namido bond lengths are longer for the high spin (HS) complexes compared to the low spin (LS) ones.| Complex | Average Fe(III)–Namido distance/Å | Number of Fe(III)–Namido bonds | Spin state of iron(III) | Reference |
|---|---|---|---|---|
| [Fe(Pypep)2]+ | 1.957(4) | 2 | LS | 3 |
| [Fe(Prpep)2]+ | 1.955(2) | 2 | LS | 4 |
| [Fe(PypepS)2]− | 1.954(2) | 2 | LS | 5 |
| [Fe(PypepO)2]− | 2.064(4) | 2 | HS | 21 |
| [Fe(bpc)(MeCO2)2]− | 2.064(2) | 2 | HS | 9 |
| [Fe(bpc)(1-MeIm)2]+ | 1.886(4) | 2 | LS | 10 |
| [Fe(PypepS2)]− | 2.040(3) | 2 | HS | 6 |
| [Fe(POPY)(1-MeIm)2]− | 2.228(4) | 2 | HS | 8 |
| [Fe(POPY)(NCS)2]3− | 2.224(6) | 2 | HS | 8 |
| [Fe(L)2]− | 1.971(3) | 4 | LS | 11 |
| [Fe(Py3P)2]− | 1.962(2) | 4 | LS | 7 |
| [Fe(MePy3P)2]− | 1.955(3) | 4 | LS | 7 |
| [Fe(H2O)(3)]− | 1.877(8) | 4 | IS | 12 |
| [Fe(η4-MAC*)(Cl)]2− | 1.927(16) | 4 | IS | 14 |
3.1 Complexes with two carboxamido nitrogens
The bis complexes of the tridentate ligands PypepH and PrpepH were the first examples of structurally characterized Fe(III) complexes that contain ligated carboxamido nitrogens.3,4 The deprotonated ligands, Pypep− and Prpep−, form octahedral Fe(III) complexes with the two carboxamido nitrogens trans to each other (Fig. 2a and 2b). The complexes are thus the mer isomers. The average Fe(III)–Namido bond distances in [Fe(Pypep)2]Cl and [Fe(Prpep)2]ClO4 are 1.957(4) Å and 1.955(2) Å respectively.3,4 Comparison of the Fe–Namido distances in [Fe(Prpep)2] (1.984(6) Å) and [Fe(Prpep)2]ClO4 reveals that carboxamido nitrogen donors favor the Fe(III) center over Fe(II). This fact has been discussed in detail in our previous paper.4 The average Fe(III)–Namido bond distance in the two bis complexes [Fe(PypepO)2]− and [Fe(PypepS)2]− (Fig. 2c and 2d) differs by 0.11 Å (Table 1)5,21 presumably due to a change in the spin state (HS to LS) upon replacement of the phenolato oxygen by thiolato sulfur around the Fe(III) center. In the Fe(III) complexes of the ligand bpcH2 (Fig. 3), the carboxamido nitrogens are situated in a cis configuration. The average Fe(III)–Namido bond distance in [Fe(bpc)(MeCO2)2]− (2.064(2) Å, Fig. 3a) is considerably longer than that in [Fe(bpc)(1-MeIm)2]+ (1.886(4) Å, Fig. 3b). This large difference in bond length could be related to different spin states (HS vs. LS respectively) in addition to the overall charge of the species.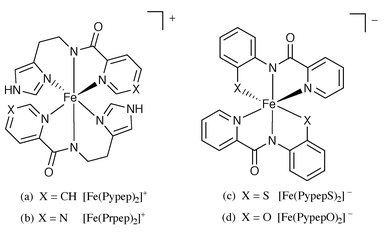 | ||
| Fig. 2 | ||
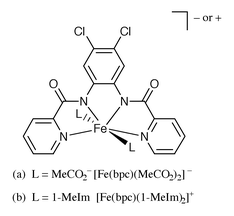 | ||
| Fig. 3 | ||
Another interesting fact emerges upon comparison of the structural parameters of [Fe(bpc)(1-MeIm)2]+ (Fig. 3b) and [Fe(Pypep)2]+ (Fig. 2a). These two complexes have identical donor atoms around the Fe(III) center. However, the average Fe(III)–Namido bond length in [Fe(bpc)(1-MeIm)2]+ is shorter than that in [Fe(Pypep)2]+ (1.886(4) Å vs. 1.957(4) Å). This difference clearly arises from a trans-effect since the carboxamido nitrogens are cis to each other in the former complex while the same nitrogens are in a trans configuration in the latter.
The two pentadentate ligands PypepS2H4 and POPYH4 afford Fe(III) complexes of different geometries. The fully deprotonated pentadentate PypepS24− ligand coordinates to Fe(III) in a helical geometry (Fig. 4a).6 The average Fe(III)–Namido distance noted with HS [Fe(PypepS2)]− is 2.040(3) Å. Quite in contrast, the fully deprotonated POPY4− assumes a planar conformation and occupies the equatorial plane of seven coordinate pentagonal bipyramid geometry (Fig. 4b) in [Fe(POPY)(X)]n− (when X = 1-MeIm, n = 1; X = NCS−, n = 3). In these two HS seven coordinate complexes, the average Fe(III)–Namido distances are virtually identical (Table 1) despite very different overall charges.
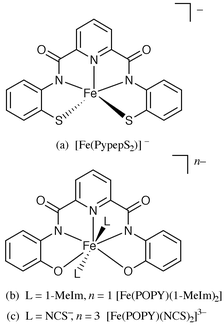 | ||
| Fig. 4 | ||
3.2 Complexes with four carboxamido nitrogens
In all the structurally characterized Fe(III) complexes, the four carboxamido nitrogens are situated in the equatorial plane. In [Fe(L)2]−, the Fe(III) center is coordinated to the Namide–Npyridine–Namide frame of the two ligands in mer fashion (Fig. 5a).11 Similar coordination is observed in [Fe(Py3P)2]− and [Fe(MePy3P)2]− (Fig. 5b and 5c) despite the presence of two pendant pyridine arms in the ligand frames.6 It thus becomes evident that in all three species, the Namide–Npyridine–Namide portion of the ligands provide exceptional stability to the Fe(III) center. The enhanced stability is further supported by the fact that only the bis complexes [Fe(Py3P)2]− and [Fe(MePy3P)2]− are isolated even when the reaction mixtures contain 1∶1 metal-to-ligand ratios. Clearly, enhanced stability provided by the carboxamido nitrogen donors outweighs the chelate effect of the pentadentate ligands in the latter two complexes. The average Fe(III)–Namido bond distances in all three complexes are very similar to each other (Table 1).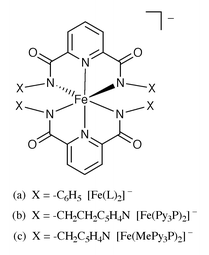 | ||
| Fig. 5 | ||
There are two other examples of Fe(III) complexes with four carboxamido nitrogens. These are derived from the macrocyclic ligands H4[MAC*] and H4[3].12–14 The fully deprotonated tetraanionic ligands occupy the equatorial plane in both complexes with distorted square pyramidal geometry (Fig. 6a and 6b). The Fe(III)–Namido bond distances are shorter in these intermediate spin (IS) species (1.927(3) and 1.877(8) Å for and [Fe(η4-MAC*)(Cl)]2− and [Fe(H2O)[3]]− respectively) presumably due to the structural constraints of the macrocyclic ligand frames.
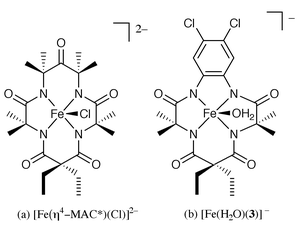 | ||
| Fig. 6 | ||
4 Spectroscopic and redox properties
Ligation of carboxamido nitrogen to Fe(III) centers can be conveniently followed by (a) disappearance of the N–H stretch(es) and (b) shift of the strong carbonyl stretching band (νCO) to lower frequencies. For example, the bis complex [Fe(Py3P)2]− exhibits its νCO at 1593 cm−1 while for the free ligand Py3PH2, νCO is noted at 1654 cm−1. The position of νCO provides additional information in some cases. Comparison of the νCO of the two bis complexes [Fe(Prpep)2] and [Fe(Prpep)2]+ (1590 cm−1 and 1630 cm−1 respectively relative to 1665 cm−1 in the free ligand PrpepH) reveals that the carboxamido nitrogen binds to Fe(III) more strongly than Fe(II).4 It is evident that between the two possible resonance structures shown below, the Fe(II) complex has a larger contribution of structure b while structure a contributes more to the Fe(III) complex.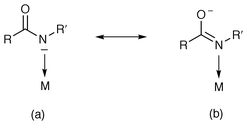
Most of the Fe(III) complexes included in this account are low spin (LS), although some high spin (HS) cases are noted (Table 1). The LS bis complexes with four carboxamido nitrogen donors in the equatorial plane (Fig. 5) exhibit sharp axial signals with g⊥ = 2.18 and g∥ = 1.94 (Table 2). Rhombic signals are usually observed for LS bis complexes with two carboxamido nitrogens like [Fe(PypepS)2]− (g = 2.22, 2.14 and 1.98) due to the inequivalent electronic axes at the metal center. The LS nature of these Fe(III) complexes appears to be a result of the strong donor capacity of the carboxamido nitrogens.
| Complex | g values | μB | E1/2 |
|---|---|---|---|
| [Fe(Pypep)2]+ | 2.22 (298K) | −0.31 (DMF) | |
| [Fe(Prpep)2]+ | 2.24 (298K) | −0.10 (DMF) | |
| [Fe(PypepS)2]− | 2.22, 2.14, 1.98 | −1.12 (DMF) | |
| [Fe(PypepO)2]− | 9.3, 4.2 | −1.08 (DMF) | |
| [Fe(bpc)(MeCO2)2]− | 5.22, 2.02 | 5.99 (300K) | −0.44 (CH3CN) |
| [Fe(bpc)(1-MeIm)2]+ | 1.79 (298K) | −0.54 (CH3CN) | |
| [Fe(PypepS2)]− | 9.3, 4.2 | 6.13 (298K) | −0.65 (DMF) |
| [Fe(POPY)(1- MeIm)2]− | 6.43, 5.55, 1.99 | −1.01 (DMF) | |
| [Fe(POPY)(NCS)2]3− | 6.74, 5.10, 2.00 | ||
| [Fe(L)2]− | 2.160, 1.990 | 2.38 (300K) | −0.91 (CH3CN) |
| [Fe(Py3P)2]− | 2.18, 1.94 | −0.95 (DMF) | |
| [Fe(MePy3P)2]− | 2.18, 1.94 | −1.05 (DMF) | |
| [Fe(H2O)[3]]− | 5.8, 5.0, 2.9, 1.8 | ||
| [Fe(η4-MAC*)(Cl)]2− | 4.38, 3.73, 2.06 |
The Fe(III) complexes of PypepS2H4 and POPYH4 with coordination number 5 and 7 are HS despite the presence of strong donors (Table 1). The unusual coordination geometries of these species (Fig. 4a–c) could be responsible for this behavior. Intermediate spin (3/2) states have also been noted with the 5 coordinate Fe(III) complexes with macrocyclic ligands (Fig. 1, Table 1). Collins and coworkers have studied the magnetic and Mössbauer properties of these species in detail.12–14
Changes in donor sets in structurally similar complexes also bring about changes in the spin state. Two interesting pairs of examples are given in Table 1. In the first case which involves the two bis complexes [Fe(PypepS)2]− and [Fe(PypepO)2]− (Fig. 2b and 2c), a change of thiolato sulfur to phenolato oxygen around Fe(III) causes a spin change from LS to HS. In the second example, a change of the neutral 1-MeIm axial donors of the LS complex [Fe(bpc)(1-MeIm)2]+ to acetate results in the HS species [Fe(bpc)(MeCO2)2]−. Clearly, several factors dictate the overall spin state of these Fe(III) complexes and more work is required to determine the contribution of the individual factors.
The half-wave potentials (E1/2) for most of the complexes have been recorded and are listed in Table 2. That the carboxamido nitrogens provide extra stability to the +3 oxidation state of iron is readily noted in the highly negative reduction potentials (≡ −1.0 V vs. SCE) for complexes like [Fe(L)2]−, [Fe(Py3P)2]−, and [Fe(MePy3P)2]− (Table 2). These complexes comprise four carboxamido and two aromatic nitrogens around the Fe(III) center. The reduction potentials drop sharply when the number of carboxamido nitrogens (negatively-charged) is decreased. For example, the bis complexes [Fe(Pypep)2]+ and [Fe(Prpep)2]+ are reduced at −0.31 V and −0.10 V vs. SCE respectively. The major part of the stability of the Fe(III) center arises from electrostatic effects of the negatively-charged donors. This is further evidenced by the reduction potentials of the complexes [Fe(PypepO)2]− and [Fe(PypepS)2]− (−1.08 and −1.12 V vs. SCE respectively) compared to [Fe(Pypep)2]+. Collectively, the redox potentials now suggest that carboxamido nitrogen stabilizes the Fe(III) center to a great extent much like the carboxylates and phenolates.23,24
5 Stability
The topic of whether the Fe(III)–Namido bond will survive in an aqueous medium has long been the subject of intense debate. Since the carboxamido nitrogen is highly basic, it was assumed to be susceptible to hydrolysis. However, the Fe(III) complexes with Fe(III)–Namido bonds are often stable in water. For example, the bis complexes [Fe(Py3P)2]− and [Fe(MePy3P)2]− are very stable in water and undergo no further reaction in the presence of strong ligands such as CN− and N3−.7 Several complexes with Fe(III)–Namido bonds have been synthesized in aqueous alcohol.3,10 In aqueous solution, [Fe(POPY)(1-MeIm)2]− exchanges 1-MeIm ligands with water reversibly without any decomposition.8 It is therefore clear that the Fe(III)–Namido bond in such complexes remains intact even during substitution reactions in water. Results of extensive spectroscopic studies on [Fe(bpc)(MeCO2)2]− also indicate that substitution of the axial acetates with a wide variety of neutral or anionic ligands does not destroy the complexes.9 In addition, the Fe(III) complexes derived from the macrocyclic ligands (Fig. 6a and 6b) have been oxidized to their Fe(IV) analogues in aqueous environments. The structurally characterized Fe(IV) species demonstrate that the Fe–Namido bonds remain intact during oxidation of the iron center.13,14 These results lend further support to our conclusion7 that the Fe(III)–Namido bond is quite stable toward hydrolytic decomposition and aqueous solutions of Fe(III) complexes with Fe(III)–Namido bonds seldom afford oxo- or hydroxo-bridged dimeric (or polymeric) Fe(III) species which are often recognized as thermodynamically very stable entities.26 Summary
Over the past few years, a new area of coordination chemistry, namely, ligation of carboxamido nitrogen to Fe(III) has begun to unfold. Results from a few laboratories including ours have provided insight into the syntheses, structures, and properties of Fe(III) complexes with one or more Fe(III)–Namido bond(s). In this account we demonstrate that these complexes are not anomalies but very stable entities in most cases. Also along this line, we emphasize that the carboxamido nitrogens are good donors for Fe(III) and impart significant stability to the Fe(III) center. We expect that this review will serve as a starting point for the preparation and characterization of additional complexes of this type in the future.7 References
References
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385 CrossRef CAS.
- S. J. Lippard, Angew. Chem., Int. Ed. Engl., 1988, 27, 344 CrossRef.
- X. Tao, D. W. Stephan and P. K. Mascharak, Inorg. Chem., 1987, 26, 754 CrossRef CAS.
- S. J. Brown, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1990, 29, 3229 CrossRef CAS.
- J. C. Noveron, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1998, 37, 1138 CrossRef CAS.
- J. C. Noveron, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., submitted..
- D. S. Marlin, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1999, 38, 3258 CrossRef CAS.
- D. S. Marlin, M. M. Olmstead and P. K. Mascharak, Inorg. Chim. Acta, in the press. Search PubMed.
- M. Ray, R. Mukherjee, J. F. Richardson and R. M. Buchanan, J. Chem. Soc., Dalton Trans., 1993, 2457 Search PubMed.
- C.-M. Che, W.-H. Leung, C.-K. Li, H.-Y. Cheng and S.-M. Peng, Inorg. Chim. Acta, 1992, 196, 43 CrossRef CAS.
- M. Ray, D. Ghosh, Z. Shirin and R. Mukherjee, Inorg. Chem., 1997, 36, 3568 CrossRef CAS.
- M. J. Bartos, C. Kidwell, K. E. Kauffmann, S. W. Gordon-Wylie, T. J. Collins, G. C. Clark, E. Münck and S. T. Wientraub, Angew. Chem., Int. Ed. Engl., 1995, 34, 1216 CrossRef CAS.
- M. J. Bartos, S. W. Gordon-Wylie, B. G. Fox, L. J. Wright, S. T. Wientraub, K. E. Kauffmann, E. Münck, K. L. Kostka, E. S. Uffelman, C. E. F. Rickard, K. R. Noon and T. J. Collins, J. Coord. Chem. Rev., 1998, 174, 361 Search PubMed.
- K. L. Kostka, B. G. Fox, M. P. Hendrich, T. J. Collins, C. E. F. Rickard, L. J. Wright and E. Münck, J. Am. Chem. Soc., 1993, 115, 6746 CrossRef CAS.
- M. Ray, A. P. Golombek, M. P. Hendrich, V. G. Young, Jr. and A. S. Borovik, J. Am. Chem. Soc., 1996, 118, 6084 CrossRef CAS.
- J. W. Peters, M. H. Stowell, M. Soltis, M. G. Finnegan, M. K. Johnson and D. C. Rees, Biochemistry, 1997, 36, 1181 CrossRef CAS.
- S. Nagashima, M. Nakasako, N. Dohmae, M. Tsujimura, K. Takio, M. Odaka, M. Yohda, N. Kamiya and I. Endo, Nat. Struct. Biol., 1998, 5, 347 CrossRef CAS.
- W. Huang, J. Jia, J. Cummings, M. Nelson, G. Schneider and Y. Lindqvist, Structure, 1997, 5, 691 CrossRef CAS.
- R. J. Guajardo, S. E. Hudson, S. J. Brown and P. K. Mascharak, J. Am. Chem. Soc., 1993, 115, 7971 CrossRef CAS.
- Y. Sugiura, T. Takita and H. Umezawa, Met. Ions Biol. Syst., 1985, 19, 81 Search PubMed.
- D. S. Marlin, M. M. Olmstead and P. K. Mascharak, unpublished work..
- L. A. Tyler, J. C. Noveron, M. M. Olmstead and P. K Mascharak, Inorg. Chem., 1999, 38, 616 CrossRef CAS.
- K. Ramesh and R. Mukherjee, J. Chem. Soc., Dalton Trans., 1992, 83 RSC.
- C. Stockheim, L. Hoster, T. Weyhermüller, K. Wieghardt and B. Nuber, J. Chem. Soc., Dalton Trans., 1996, 4409 RSC.
Footnote |
| † Ligand abbreviations used in this paper: H2L = 2,6-bis(N-phenylcarbamoyl)pyridine; Py3PH2 = N,N′-bis[2-(pyridyl)ethyl]pyridine-2,6-dicarboxamide; MePy3PH2 = N,N′-bis[2-(2-pyridyl)methyl]pyridine-2,6- dicarboxamide; H2bpc = 4,5-dichloro-1,2-bis(pyridine-2-carboxamido)- benzene; PypepH = N-[2-(4-imidazoyl)ethyl]pyridine-2-carboxamide; PrpepH = N-[2-(4-imidazolyl)ethyl]pyrimidine-4-carboxamide; PypepSH2 = N-2-mercaptophenylpyridine-2′-carboxamide; PypepOH2 = N-2-hydroxyphenylpyridine-2′-carboxamide; H4[MAC*] = 1,4,8,11-tetraaza-13,13-diethyl-2,2,5,5,7,7,10,10-octamethyl-3,6,9,12,14-p entaoxocyclotetradecane; H4[3] = 13,14-dichloro-6,6-diethyl-2,5,7,10(6H,11H)- tetraoxo-3,3,9,9-tetramethyl-1H-1,4,8,11-benzotetraazacyclotridec ine; PypepS2H4 = N′-bis(2-mercaptophenyl)pyridine-2,6-dicarboxamide; POPYH4 = N,N′-bis(2-hydroxyphenyl)pyridine-2,6-dicarboxamide. |
| This journal is © The Royal Society of Chemistry 2000 |