Photodimerization of anthracenes in fluid solution: structural aspects
Henri Bouas-Laurenta, Alain Castellanb, Jean-Pierre Desvergnea and René Lapouyadec
aLaboratoire de Photochimie Organique, LCOO, CNRS, UMR 5802, Université Bordeaux 1, 33405, Talence Cedex, France.. E-mail: jp.desvergne@lcoo.u-bordeaux.fr
bLaboratoire de Chimie des Substances
Végétales, Institut du Pin, Université Bordeaux 1, 33405, Talence Cedex, France.. E-mail: a.castellan@ipin.u-bordeaux.fr
cLaboratoire d’Analyse Chimique par Reconnaissance
Moléculaire, LACReM, Ecole Nationale Supérieure de Chimie et de Physique de
Bordeaux (ENSCPB), Talence, France. E-mail: lapouyad@enscpb.u-bordeaux.fr
First published on UnassignedUnassigned11th January 2000
Abstract
Owing to their versatile photophysical and photochemical properties, anthracene and its derivatives are being employed in many systems, for instance as energy migration probes in polymers, triplet sensitizers, molecular fluorosensors, electron acceptor or donor chromophores in artificial photosynthesis, photochromic substrates in 3D memory materials, etc. One remarkable feature is their ability to photodimerize under UV irradiation which induces considerable changes in their physical properties. This account reports the preparative and structural aspects of this very useful and protean photocycloaddition.
 Henri Bouas-Laurent Henri Bouas-Laurent | Henri Bouas-Laurent worked with Professor Raymond Calas for his thesis (1964) and was appointed Maître de Conférences in 1965 and promoted full Professor in 1970 at University Bordeaux 1, where he has been Professor Emeritus since 1998. He founded the organic photochemistry group which joined Professor Joussot-Dubien’s laboratory in 1974, together with R. Lapouyade, A. Castellan and J.-P. Desvergne. His research interests focus on the photochemistry of aromatic hydrocarbons, the photophysics of bichromophoric interactions, photochromic compounds, fluorosensors, photoactive supramolecular systems and, recently, the gelation of organic liquids with small molecules. He was co-editor of the book ‘Photochromism, molecules and systems’ with Professor H. Dürr in 1990. He is the recipient of the CNRS bronze medal (1957), the Grammaticakis-Neuman award of the French Academy of Sciences (1986), the Alexander-von-Humboldt Research prize (1991) and the Doctor h.c. degree of the University of Saarbrücken (Germany, 1999). |
 Alain Castellan Alain Castellan | Alain Castellan was born in Montceau les Mines, France in 1946, and received his thesis in 1974 from the University of Bordeaux on the ‘Photodimerisation of aromatic hydrocarbons in solution’. He spent one year (1975–1976) at the University of Utah in the laboratory of Professor Josef Michl working on the photoreactivity of polycyclobutenes from upper excited states and on magnetic circular dichroism spectroscopy. In 1985, he earned a full Professor position at University Bordeaux 1 in organic chemistry and for two years has been the Director of the ‘Laboratoire de Chimie des Substances Végétales’ at the ‘Institut du Pin’. His current research focuses on the chemistry and photochemistry of lignocellulosics in relation to wood and paper science. |
 Jean-Pierre Desvergne Jean-Pierre Desvergne | Jean-Pierre Desvergne obtained his thesis degree from the University of Bordeaux in 1973, after spending one year (Leverhulme visiting fellowship) in Aberystwyth (Wales) with Professor John M. Thomas to study the surface chemistry and photochemistry of organic crystals. He has been a member of CNRS since 1970 and is currently Director of Research at Université Bordeaux 1. He has been recently appointed head of the “Laboratoire de Chimie Organique et Organométallique” (LCOO), a mixed CNRS and University Unit. His domains of expertise and research interests span solid state molecular photochemistry, the photochemistry and photophysics of aromatic hydrocarbons, polymer photochemistry, molecular and cation recognition, as well as structural and spectroscopic studies of gels of organic liquids. He was co-editor of the book ‘Chemosensors of Ion and Molecule Recognition’ with Professor A. W. Czarnik, in 1997. |
 René Lapouyade René Lapouyade | René Lapouyade was born in Saint Germain des Prés, Dordogne, France. He obtained his thesis degree in 1969 with Professor H. Bouas-Laurent at the University of Bordeaux. For this work on the ‘Peri effect and photochemical reactivity of anthracene derivatives’ he received the Adrian award from the Societe Chimique de France. After one year of postdoctoral research at the Photochemistry Unit of the Western Ontario University, Canada, with Professor Paul de Mayo, he joined the photochemistry group of Bordeaux headed by Professor Jacques Joussot-Dubien. He was named CNRS Research Director in 1976. He participated in the creation of a new research group in the Institut de Chimie de la Matière Condensée de Bordeaux (ICMCB) from 1994 to 1998 and is now working in the Laboratory of Chemical Analysis from Molecular Recognition (LACReM) in the Ecole Nationale Supérieure de Chimie et Physique de Bordeaux (ENSCPB). His research interests include synthesis and photophysical studies of new supramolecular fluorophores for the selective recognition of ions and molecules. Currently he is engaged in the design of new photochromic ionophores to detect ions and achieve fast ion concentration jumps and spin transitions. |
1 Introduction
Over the last sixty years, a number of polycyclic aromatic hydrocarbons (PAH) have become notorious as pollutants. Research in this field has been therefore stimulated, especially in the development of analytical methods to detect traces; these involve, inter alia, their efficient light emission properties. Among PAH, anthracene and its derivatives have been extensively investigated; this is reflected in the ca. 38 000 publications dealing with anthracenes between 1967 and 1998; about 4000 of them were devoted to the photochemical reactivity (Chemical Abstracts).Of special interest are the bimolecular photochemical reactions of anthracenes. The ring is liable to act as a light induced electron donor or acceptor, a property easily tuned by substitution. Anthracenes also possess photochromic properties which can be used in the design of optical, electronic or magnetic switches incorporated in mesophases, polymers, films or crystals. These reversible properties are based on the photodimerization reaction. This account is limited to the fundamental aspects of the latter reaction in fluid solutions. Several reviews on the photophysics and photochemistry of anthracenes have been published1,2 but none of them covers all the structural aspects of the photodimerization.
2 Brief historical background: the pioneering period
Anthracene photodimerization is one of the oldest known photochemical reactions. Fritzsche in 18671–3 discovered that solar irradiation of a saturated benzenic solution of a hydrocarbon (later named ‘photene’) leads to ‘microscopic crystals which adhere firmly to the sides of the vessel’; they were found to be inert and almost insoluble in organic solvents; remarkably, the photoproduct, called paraphotene in 1869, was observed to revert back entirely to the starting compound thermally (slowly at room temperature but faster at 170 °C in phenetole).1,3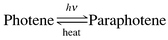 | (1) |
But the exact structure was not determined with certitude until the first crude X-ray analysis by Hengstenberg in 1932.5 Further evidence of the 9,9′ and 10,10′ bonding was provided by Coulson in 1955, using UV spectrometry.1 The X-ray structure was confirmed by Ehrenberg in 1966.6
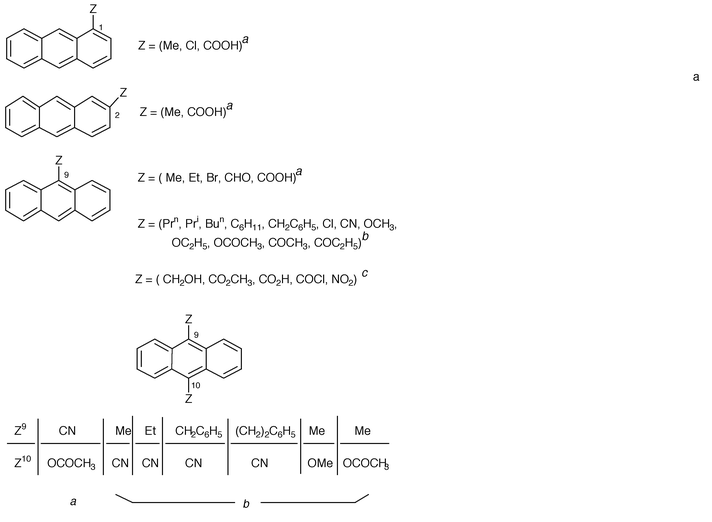
During the 1900–1950 period, the photoreaction was sporadically extended to some monosubstituted derivatives (CH3, C2H5, Cl, Br, CHO, COOH) in the 1, 2 or 9 position and to one 9,10-disubstituted substrate (CN, OCOCH3) as reported by Calas and Lalande (1960),1,4† see Table 1. The latter achieved the first systematic investigation of the photodimerization of anthracene derivatives, substituted in the meso position (Table 1). In parallel, Applequist (1959)1 and Greene (1960)1 prepared some selected photodimers as starting materials for the then hypothetical 9,10-dehydrodianthracene1 and, along with Calas and Lalande (1960),1,4 tackled the structural problems of 9-substituted anthracene photodimers. Indeed, the two monomers can theoretically associate with a head-to-head (hh, also termed cis) or a head-to-tail (ht, also termed trans) mutual orientation, leading to the hh and ht photodimers represented in Scheme 1.
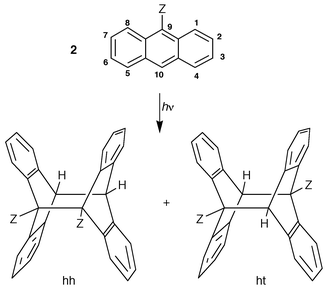 | ||
| Scheme 1 Photodimerization of 9-substituted anthracene derivatives may lead to head-to-head (hh) and head-to-tail (ht) photodimers. | ||
By measuring the dipole moments of the isolated photodimers of 9-bromo- and 9-cyanoanthracenes, Calas et al. demonstrated that their structure was ht.4 Independently, Applequist (1959)1 arrived at the same conclusion by applying the same technique to the 9-chloro, 9-formyl and 9-chlorocarbonyl derivatives (vide infra).
The photodimers were prepared according to the following procedure: an aerated (non degassed) solution of the monomer at concentration 5 × 10−2 M to 10−1 M in an organic solvent (benzene, cyclohexane, ether, THF, methanol, ethanol) was irradiated for about 24 hours in a Pyrex vessel with an external high pressure mercury lamp which, in addition to its photochemical effect, provides enough heat to maintain the medium under gentle reflux. Owing to their poor solubility, the photoproducts were observed to precipitate, or stick to the reactor’s walls. After solvent evaporation, the solid was washed to eliminate the non reacted monomer and recrystallized in benzene (for instance, 1 litre of benzene is necessary to recrystallise ca. 400 mg of dianthracene, one of the least soluble).4
In 1960, Calas and Lalande concluded their study with the following statement:4 by UV irradiation in solution, most anthracene derivatives lead to colourless photodimers, not fluorescent in daylight, and poorly soluble in organic solvents. The photodimers were shown to thermally regenerate the monomers. From the demonstration of the ht structure of some of the 9-substituted photodimers, the authors inferred, considering steric and electronic factors, the generality of this structure. Moreover, they discussed their thermal stability by considering the difference of melting point between dimer and monomer (Calas and Lalande).4
These systematic investigations of simple substituent effects on the structure of the photoproducts stimulated the exploration of the scope of the reaction and in parallel, the first developments of mechanistic studies.
3 Expansion of the field: scope of the reaction
3.1 Intermolecular photodimerization
3.1.1.1 9-Monosubstituted anthracenes.. From the fifties up to now, a number of other derivatives (a selection has been listed in Table 2) have been photodimerized in order to explore the scope of the reaction and establish the still unknown structures. Although most derivatives can photodimerize in a variety of organic solvents, it was shown that some solvents could affect the photoreaction: thus 9-formylanthracene does photodimerize in benzene or toluene but not in ethanol where it undergoes reduction.7 It was also found by Bowen (1953,1955)1 that heavy atom solvents, such as CCl4, CS2 and C6H5Br are detrimental. 9-Nitroanthracene was shown to photodimerize only in the wavelength range 420–530 nm; in the range 300–400 nm it is photolyzed into a complex mixture of compounds (Greene, 1960).1
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) NOH
NOH![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) C–C6H5
C–C6H5Chemical correlation was extensively used as a method of structure identification before the advent of high sensitivity NMR equipment. The ht structure of 9-nitroanthracene photodimer (Scheme 2) was determined by reduction to a 9-aminoanthracene photodimer whose ht structure had been established by Chapman (1969),1 using NMR spectra. The amino derivative structure was also correlated to that of the diacid (Z = CO2H) through the formation of the COCl, CON3 and NCO derivatives.
 | ||
| Scheme 2 Chemical correlation assigning the ht structure of the isolated 9-nitroanthracene photodimer (Chapman et al., 1969).1 | ||
But structures determined with chemical methods sometimes lead to erroneous results, as shown in the following: in an attempt to obtain a hh isomer, Greene and coworkers (1960)1 irradiated 9-anthroyl anhydride to generate its photocyclomer; LiAlH4 reduction of the latter was described in 1955 as generating the hh dimer of 9-hydroxymethylanthracene (Scheme 3). However the authors observed later that, at the concentration used (4 × 10−2 M) they had obtained a polymeric ht anhydride which, on reduction, provided the ht dimer of 9-hydroxymethylanthracene. The ht structure of the 9-hydroxymethylanthracene photodimer thus obtained was established by high yield reduction of the ht photodimer of 9-anthraldehyde, whose structure had been assessed by dipole moment measurement (Scheme 3). Despite this correction published 5 years later, the first paper was often wrongly cited as evidence for the hh structure of 9-substituted anthracene photodimers. Many years later, it could be shown by NMR of the crude photoproduct that the irradiation of 9-hydroxymethylanthracene leads to a mixture of hh and ht photodimers (see section 4.6 and Table 6).
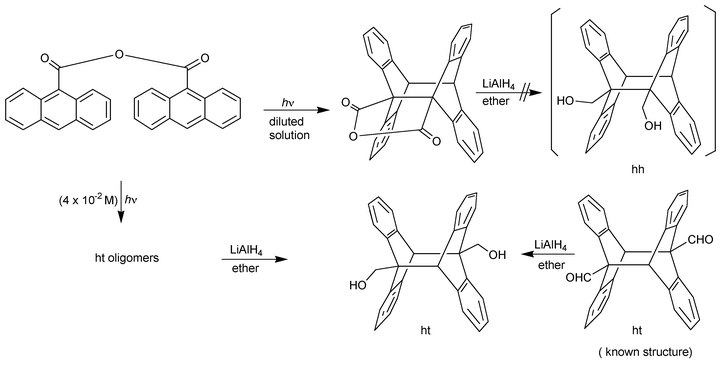 | ||
| Scheme 3 Chemical correlations to establish the structure of 9-hydroxymethylanthracene photodimer; it was wrongly published to be hh in 1955, but, after reinvestigation, demonstrated to be ht; 9-anthroyl anhydride was obtained only in dilute solution and the product of its reduction was not reported (Greene, 1960).1 | ||
At that time, the available experimental evidence strongly suggested that all photodimers should have a ht structure, all but 9-deuterioanthracene as far as deuterium was accepted as a substituent; this derivative was expected to yield a 50∶50 mixture of hh and ht photodimers. The pure ht isomer (Scheme 4) could be prepared from the 9-bromo-10-deuteriodianthracene known to have the ht structure (dipole moment measurement); treatment with a Grignard reagent led to the ht 9-deuterioanthracene dimer whose IR spectrum was compared with that of the irradiation photoproduct. That the latter exhibits four more absorption bands than the pure ht dimer can be considered as good evidence of the hh 9-deuteriodianthracene formation.15
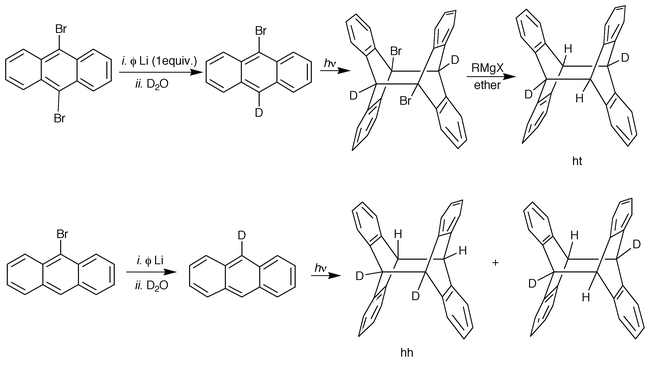 | ||
| Scheme 4 Preparation of the pure ht and of a 50∶50 hh and ht mixture of 9-deuterioanthracene photodimers. | ||
Among the 9-monosubstituted derivatives examined by Calas and Lalande (1960),4 9-phenyl- and 9-benzoylanthracenes4 were found unexpectedly not to yield their respective photodimers whereas those of 9-cyclohexyl, 9-vinyl and 9-acetyl derivatives had been characterized. However, the 9-phenylanthracene and 9-benzoylanthracene classical photodimers were isolated by Kaupp et al. (1980),11 and Becker,12 respectively; the previously reported negative experiments might be due to the presence of quenching impurities.
3.1.1.2 Anthracenes monosubstituted in other positions.. 1- or 2-Monosubstituted anthracenes are conducive to photodimerization (Table 1). In this case, not two but four photodimers are to be expected, which may be difficult to separate. Such a separation has been accomplished by chromatography for 1-chloroanthracene (Heller and Schmidt, 1971),14 2-anthrylsulfonic acid (Tamaki et al., 1987),16 1-anthrylcarboxylic acid (Ueno et al., 1991)16 and 2-anthryloxy derivatives (Lin et al., 1989).16
Moreover, Becker has recently isolated, in addition to the above mentioned four dimers a dissymmetrical ht photodimer involving the 9,10- and 1′,4′-positions for 1-acetylanthracene16 and methyl 1-anthrylcarboxylate.16
3.1.1.3 9,10-Disubstituted anthracenes.. Only two symmetrical derivatives are known to photodimerize: 9,10-dimethylanthracene (Bouas-Laurent et al., 1970)1 and 9,10-difluoroanthracene.11 Several dissymmetrical derivatives have been among the first to undergo photodimerization (Table 1); the photoproducts were found to be thermally labile (Calas, 1960)4 when one of the substituents, at least, is an electron-attractive group (CN, OAc); 9-cyano-10-methoxyanthracene also leads to an unstable photodimer (Castellan et al., 1975).1 The 9-methyl-10-methoxyanthracene dimer was found to be a little more stable (Calas and Lalande, 1960,1,4 Becker14).
3.1.1.4 Disubstituted or trisubstituted anthracenes in positions other than 9,10.. The photochemistry of some x,y-dichloroanthracenes has been explored by Desvergne et al. (1978).16 Particularly investigated was a series of x,y-didecyloxyanthracenes (x,y = 1,4; 1,5; 1,8; 2,6) which have been found by Brotin et al. (1992)1,16 to photodimerize. An exceptional behaviour was noted for the 2,6 derivative by Fages et al., (1988),16Scheme 6. The photodimerization of several trisubstituted derivatives has been reported by Lapouyade and coworkers.17
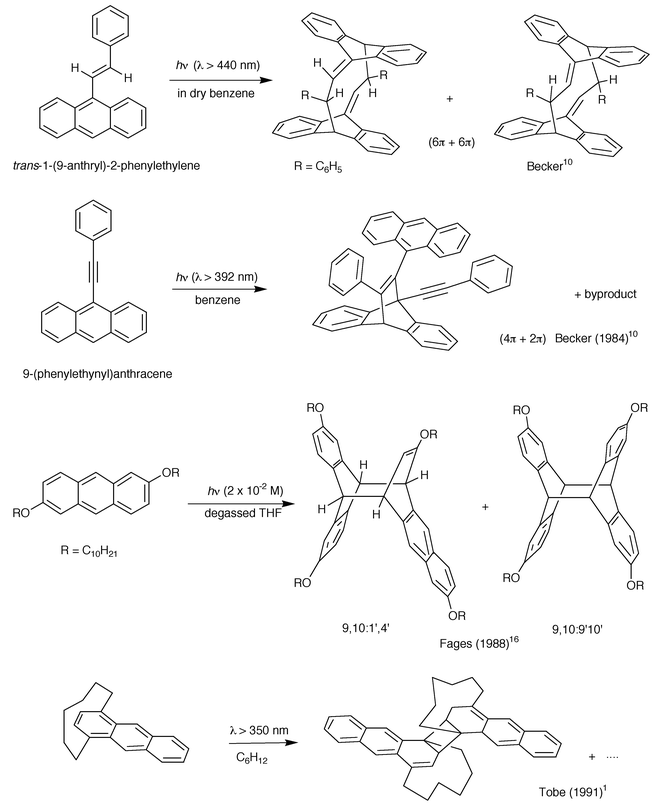 | ||
| Scheme 6 Non classical photodimers of anthracene derivatives: (6π + 6π), (4π + 2π), (4π + 4π / 9,10∶1′,4′) and (2π + 2π) cycloaddition. | ||
3.1.1.5 Failures.. Despite the fact that 9-bromoanthracene readily photodimerizes, it was shown to partially undergo photolysis of the C–Br bond (Bouas-Laurent, 1964).1 This is presumably one reason (the other being electronic) why 1-cyano-10-bromoanthracene was not observed to undergo photodimerization (Bouas-Laurent, 1964).1 Most symmetrical 9,10-disubstituted anthracenes failed to photodimerize: dialkyl (R
![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) Et), Ph, OMe (OR), Cl, Br, CN;
9,10-diphenylanthracene does not even form an excimer (for steric reasons)
and is used as a standard for fluorescence quantum yield measurements
(Birks, 1970).1,4 For 9-alkyl
substituents, the boundary between photodimerization ability and lack of
photodimerization lies between isopropyl and tert-butyl. The
latter was found by Gusten (1980)1 and
Dreeskamp (1981)1 to convert into the
9,10-Dewar isomer. However steric inhibition of photodimerization induces
an acceleration for the (4 + 2) reaction with singlet oxygen17 (Scheme 5).
Conversely 9-trimethylsilyl-anthracene was observed to smoothly
photodimerize in non protic solvents (Desvergne et al.,
1986)1 on account of the longer C9–Si
distance which relieves the steric crowding at the reaction centre.
Et), Ph, OMe (OR), Cl, Br, CN;
9,10-diphenylanthracene does not even form an excimer (for steric reasons)
and is used as a standard for fluorescence quantum yield measurements
(Birks, 1970).1,4 For 9-alkyl
substituents, the boundary between photodimerization ability and lack of
photodimerization lies between isopropyl and tert-butyl. The
latter was found by Gusten (1980)1 and
Dreeskamp (1981)1 to convert into the
9,10-Dewar isomer. However steric inhibition of photodimerization induces
an acceleration for the (4 + 2) reaction with singlet oxygen17 (Scheme 5).
Conversely 9-trimethylsilyl-anthracene was observed to smoothly
photodimerize in non protic solvents (Desvergne et al.,
1986)1 on account of the longer C9–Si
distance which relieves the steric crowding at the reaction centre.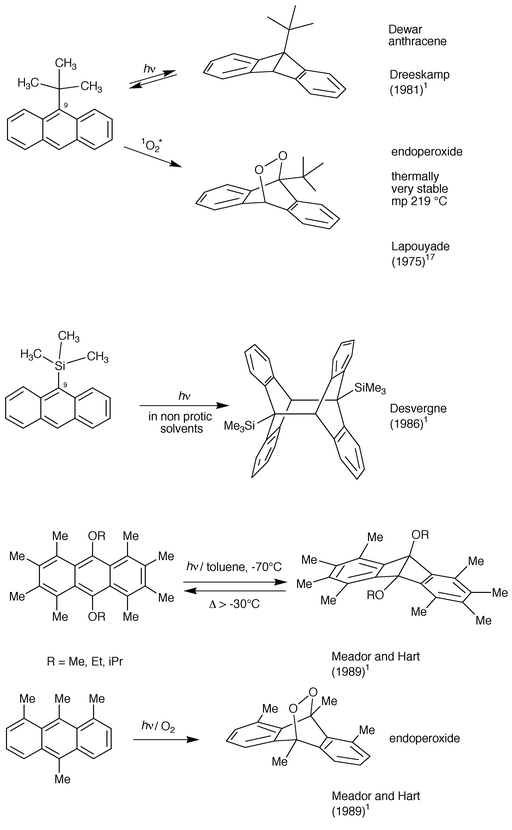 | ||
| Scheme 5 Photochemical reactions of sterically crowded meso-substituted anthracenes. | ||
Such a steric hindrance in other crowded anthracenes was also found by Meador and Hart (1989)1 to preclude photodimerization and favour Dewar anthracene or photooxide (= endoperoxide) formation (Scheme 5).
3.1.1.6 Non classical photodimers.. Although 9-vinylanthracene leads to its (4π + 4π) 9,10∶9′,10′ photodimer18 other conjugated substituents such as styryl and phenylethynyl were found to generate a (6π + 6π)10 and a (4π + 2π) (Becker, 1984)10 photocyclodimerisation respectively as the main reaction (Scheme 6). The 4π + 2π photodimer was confirmed by X-ray structure analysis (Becker, 1984).10
A dissymmetrical photodimer, formed by 9,10∶1′,4′ cycloaddition (Scheme 6), was isolated as the major photoproduct from the irradiation of 2,6-didecyloxyanthracene in degassed THF; the structure was established by UV spectrometry and NMR (Fages, 1988).16 Finally, Tobe et al., in 1991,1 discovered the (1,2∶1′,2′) photodimerization of [6](1,4)anthracenophane, an unexpected (2 + 2) dimerization in a strained anthracene. One (the major) of the five isolated photodimers is represented in Scheme 6.
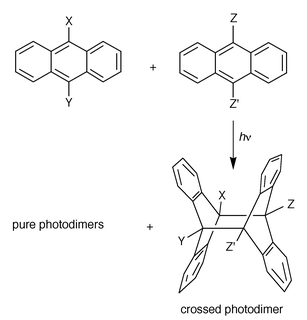 | ||
| Scheme 7 Crossed (mixed) photodimerization liable to produce a mixture of crossed and pure photodimers (see Table 3); theoretically, two crossed photodimers are possible only in the case of footnote f, in Table 3, where the hh photoproduct was obtained. | ||
The first crossed photodimers (X = Y = H, Z = Me, Bun, Cl, Br; Z′ = H, etc.) were prepared by Applequist et al. (1964)20 to serve as starting materials for the synthesis of the then elusive 9,9′-dehydrodianthracene, a molecule of interest for its unique structure. The others were obtained essentially for mechanistic purposes, i.e. to test the role of exciplexes and electron transfer between donor and acceptor partners in the photodimerization. In this context, it seems relevant to note the preparation of mixed derivatives (not reported in Table 3 because they are not photodimers) between anthracene or 9,10-dimethylanthracene and benz[a]anthracene or naphthacene respectively (Bouas-Laurent et al., 1970)1 as well as 9-cyanoanthracene with 2-methylnaphthalene.19
| Entry | X | Y | Z | Z′ | Ref. |
|---|---|---|---|---|---|
| a Applequist et al. (1964), in ref. 20.b Greene (1960), in refs. 1 and 20.c Vember (1966), in refs. 1 and 20.d Ref. 19.e Ref. 20 and Bouas-Laurent (1969), in ref. 4.f Castellan et al. (1975), in refs. 1 and 14.g Ref. 21.h Fages (1985), in ref. 1. | |||||
| 1 | H | H | Me | H | a |
| 2 | H | H | Bun | H | a |
| 3 | H | H | Cl | H | a |
| 4 | H | H | Br | H | a |
| 5 | H | H | Cl | Cl | a |
| 6 | H | H | Br | Br | a |
| 7 | H | H | CHO | H | b |
| 8 | H | H | Prn | Prn | c |
| 9 | H | H | Me | CH2OMe | c |
| 10 | H | H | CN | H | d |
| 11 | H | H | OMe | OMe | e |
| 12 | H | H | Me | Me | e |
| 13 | Cl | H | Me | Me | e |
| 14 | Br | H | Me | Me | e |
| 15 | CN | H | Me | Me | e |
| 16 | CN | H | OMe | H | f |
| 17 | Me | Me | OMe | OMe | g |
| 18 | H | H | OR | OR | h |
| 19 | H | H | R | R | h |
Table 3 suggests the following comments: (a) 9,10-dibromodianthracene could not be obtained photochemically by Applequist but was prepared by an interesting chemical transformation from 9,10-dichlorodianthracene. (The latter was allowed to react with Ph3CNa in benzene–ether to give 9,10-dehydrodianthracene (a ‘Dewar’ form of anthracene) which was found to undergo bromine addition in refluxing carbon tetrachloride (in the presence of peroxide) to yield 9,10-dibromodianthracene). (b) 9,10-Dimethyl-9′,10′-dimethoxydianthracene (Scheme 8) is the only meso crossed tetrasubstituted derivative known. It was found to be more soluble than the centrosymmetric pure photodimer of 9-methyl-10-methoxyanthracene and to decompose thermally in benzene solution ca. 4 times faster than the pure dimer.21 (c) The irradiation of a 1∶1 mixture of 9,10-dimethylanthracene (DMA) and 9-cyanoanthracene (CNA) leads to a mixture of the mixed photodimer and pure 9-cyanoanthracene photodimer (Bouas-Laurent et al., 1969).4 Their proportion strongly depends on temperature (continuous irradiation in boiling benzene favours the pure dimer which is thermally more stable than the mixed compound) and solvent. A polar solvent such as acetonitrile inducing an electron transfer between DMA (donor) and CNA (acceptor) strongly decreases the proportion of mixed dimer. But the mixed dimer was largely predominant in ether, at room temperature. (d) Based on preceding experience on electronic, steric and donor–acceptor interactions, a hh mixed photodimer between 9-methoxyanthracene and 9-cyanoanthracene was designed and successfully prepared (Castellan et al., 1975).1,14 The selectivity again strongly depends on solvent and temperature, the hh compound being thermally unstable. (e) In order to increase the solubility of the photodimers in organic solvents (hence in polymers for photochromic applications), 9,10-didecyldianthracene and 9,10-didecyloxydianthracene were prepared by Fages et al. (1985).1 Their solubility in non protic solvents was found to be ca. 500 to 1000 times that of 9,10-dimethyldianthracene (the latter is 10−3 M in C6H6 or CCl4).
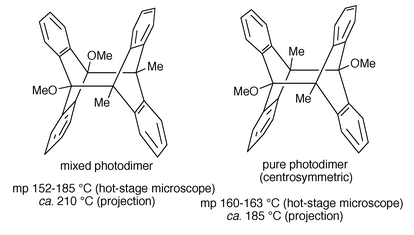 | ||
| Scheme 8 Mixed and pure meso-tetrasubstituted photodimers. | ||
3.2 Intramolecular processes∶photocycloisomerization
3.2.2.1 Non cyclic non conjugated bisanthracenes.. Most bichromophores are linked by polymethylene chains due to their transparency to light irradiation and their relative chemical inertness. Replacement of a methylene by an oxygen atom increases the flexibility (this is also the case for NH to some extent) and SiMe2 groups introduce steric constraints; other features come from functional groups which may influence not only the molecular dynamics but also the photochemistry. Let us consider successively spacers having one, two, three and more members.
One membered chain: di(9-anthryl)methane was shown by Bergmark et al., (1978)1 to photocyclize in a classical way (9,10∶9′,10′ closure) but another photocyclization mode has been observed (9,10∶1′,2′ closure) for related derivatives, arising presumably from ground state conformations conducive to (4π + 2π) cycloaddition (Scheme 9) by Becker et al. (1983, 1985, 1986, 1989)1 and Daney et al. (1985).1
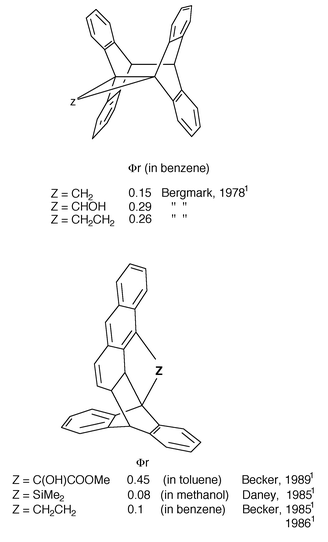 | ||
| Scheme 9 Intramolecular photocycloaddition of some bisanthracenes with one or two member spacers and their cyclization quantum yield (ϕr). | ||
Two membered chain: it is remarkable that 1,2-di(9-anthryl)ethane leads by direct irradiation (phiR: 0.26 in benzene, Bergmark et al., 1978)1 to the classical photocyclomer whereas, by sensitization with biacetyl in a benzene solution, Becker (1985)1 found a 9,10∶1′,2′ isomerization with quantum yield 0.1; thus the triplet state induces a 4π + 2π cycloaddition (Scheme 9).
Three membered chain: these short chains are known to be ideally suited for photocyclomerization; the classical (9,10∶9′,10′) cyclization has been observed to proceed efficiently for diverse linkers, through the singlet state e.g. with CH2OCH2 (Castellan et al., 1979)1 or OCH2O22 or (CH2)31,4 and the triplet state with, for instance, –CO–CH2–CH2–1 or –CO–CH(CH3)–CH2–;1 in this case, the triplet pathway is the most efficient way to the photocyclomer ever experienced (phiR: 0.65–0.72 in benzene, Becker, 1989).1 The CH2–NR–CH2 chain (R = H, CH3, CH2C6H5) was also shown to provide the corresponding photocyclomers.23 However, the SiMe2–O–SiMe2 and SiMe2–CH2–SiMe2 spacers induce a 9,10∶1′,4′ photocyclomerization as shown by Desvergne et al. (1989);1 this unusual reaction reflects the ground state conformation of the bichromophores; such a photocyclization was designed by Castellan et al. (1979)1 by linking the CH2OCH2 chain with the 9 position of one ring and the 1 position of the other anthracene (Scheme 10).
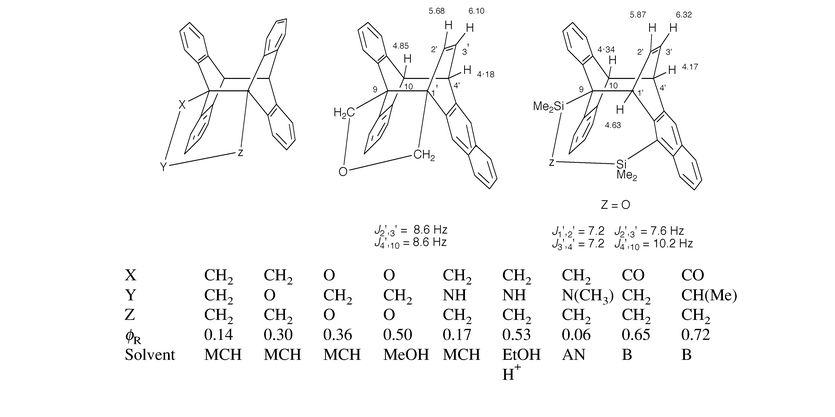 | ||
| Scheme 10 Intramolecular (4+4) photocycloaddition of some bisanthracenes with three member spacers and their cyclization quantum yield (phiR). Some relevant chemical shifts (δ, CDCl3) are given. MCH = methylcyclohexane; AN = acetonitrile: B = benzene. | ||
Chains with more than three links: most systems involve 9-anthryl terminal groups (1- or 2-anthryl groups are the exceptions) which are linked by polymethylene chains –(CH2)n– with n = 1–10 (Castellan et al., 19801) and 12, 14, 18 (Ikeda et al., 19901), polyoxyethylene (POE) sequences (Desvergne et al., 19801) such as (OCH2–CH2)n–O– with n = 3–6 and a variety of diesters: –O–CO–(CH2)2–CO–O – (Greene, 19601), –(CH2)2–O–CO–(CH2 )3–CO–O–(CH2)2 – (Fox, 19901), –CH2–O–CO–(CH2)n –O–CO–CH2– (De Schryver, 19711) with n = 7, 8, –CO–O–(CH2)n–O –CO– with n = 11, 12 for 9-anthryl groups (De Schryver, 1971)1 and n = 2, 3 for 1-anthryl groups, n = 5, 7, 9 for 2-anthryl chromophores (De Schryver, 1973)1 as well as n = 3 with 2-anthryl groups (Boens, 1976),1 see Scheme 11.
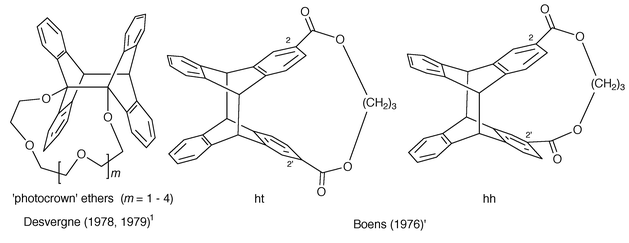 | ||
| Scheme 11 Intramolecular photocycloadducts of some bisanthracenes with spacers containing more than three members. | ||
For α,ω-(di-9-anthryl)alkanes (A–(CH2)n–A), no photocyclomers have been identified and isolated for n > 4. The photoproducts of the irradiation of A–(CH2)5–A and A–(CH2)6–A have not been characterized (Castellan, 1980).1,4 In contrast, the di-9-anthrylpolyether: A–(O–CH2–CH2)n –O–A (Desvergne et al., 1978, 1979)1 generates crown ethers (Scheme 11) by photocyclomerization; the latter is much more efficient than for the dianthrylalkanes, owing to the helicity and the great flexibility of the POE chain (the quantum yields vary in benzene from 0.26 for n = 3 to 0.20 for n = 6 i.e. for 19 atoms between the reacting centres). The presence of two ester groups close to the reacting centres in the connecting chain was found to hinder the intramolecular reaction. De Schryver and coworkers (1971)1 took advantage of this intramolecular inertness to prepare photochromic photopolymers e.g. with Mn = 52000 for A–CO–O–(CH2)11–O–CO –A involving 86 repeating units, linked in the hh and ht fashion.
3.2.2.2 Cyclic non conjugated bisanthracenes: anthracenophanes.. The relative rigidity of cyclophanes makes them particularly attractive to study the connection between the internal geometry and the fluorescence and the photoreactivity; they have been extensively studied by Ferguson (1986),1 Mataga, Misumi (1976, 1978)1 and their coworkers. [2.2](9,10) anthracenophane, first prepared as a poorly soluble yellow solid by Golden (1961)1 photoisomerizes easily in solution (phiR : 0.36) and in the solid state, even at 4 K (Scheme 12) but the photocyclomer is not thermally stable (Ferguson, 1986).1 Incidentally, it is interesting to note that the syn-[2.2](1,4)anthracenophane photoisomerizes into the 9,10∶9′,10′ cycloadduct (more reactive positions) and not through the 1,4∶1′,4′ less reactive positions (Scheme 12) although the latter are closer to each other (Morita, 1978).1
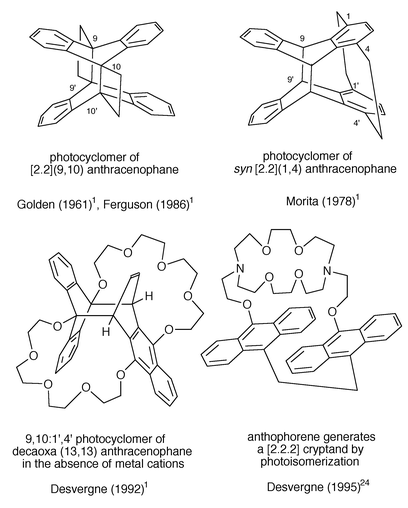 | ||
| Scheme 12 | ||
Bisanthracenes incorporated in macrocyclic metal cation receptors have been shown to display the properties of fluorosensors and cation modulated opto switches (Bouas-Laurent et al.. 1986, 1991).1 Special features are the non symmetrical photocyclomerisation of the decaoxa(13,13)anthracenophane (Desvergne et al., 1992)1 and the photocycloaddition of anthophorene (Scheme 12) into a regular cryptand.24
3.2.2.3 Receptors with two anthracenic arms.. In line with the preceding paragraph, it seems appropriate to highlight three examples of molecular or ionic receptors, different from anthracenophanes, where the anthracene photocycloaddition has been employed in order to modulate the binding ability, as illustrated in Scheme 13.
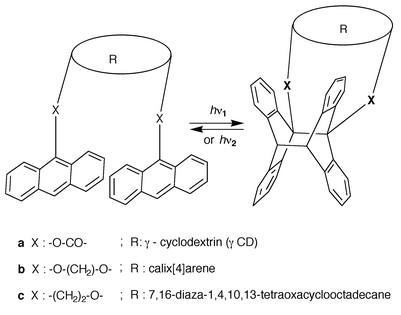 | ||
| Scheme 13 Molecular and ionic receptors whose binding ability is modified by photocycloaddition. | ||
As sketched in Scheme 13a, in the γ-CD system of Ueno et al.,25 prior to irradiation, the cavity is flexible enough to accommodate a guest (e.g. 1-borneol) but after photocycloadditon it becomes rigid and thus exhibits a very poor binding ability. Deng et al.26 (Scheme 13b) showed that the two anthracene moieties linked to calix[4]arene were transformed into a ‘lid’ by photocycloaddition; this operation, which is reversible, was shown to strongly increase the binding affinity towards Na+ as compared with the other alkali metal cations. The system (c) prepared by Tucker et al.27 is a cation modulated opto switch based on reversible light induced crown ether to cryptand transformation.
4 Physical properties and structure determination
Many photodimers and photocyclomers, particularly those with a centrosymmetric structure, are solids, poorly soluble in organic solvents. They are generally purified by recrystallization. Column chromatography can also be applied, especially for the more soluble compounds.10 Their physical properties are surveyed below.4.1 Melting points
The thermal instability of photodimers is reflected in their relatively low ‘melting points’ (often decomposition temperatures) whose values (not corrected) depend on the equipment used and the experimentalist’s skill. Kofler bench and Maquenne block allow the determination of the instantaneous decomposition (decomp.) temperature by projection whereas hot stage microscopes lead to lower temperatures because of sample preheating and therefore depend on the rate of heating. Dianthracene itself sublimes and its mp is very difficult to determine by projection (∼ 270–280 °C). Several mp’s of 9-monosubstituted and 9,10-disubstituted monomers and of their photodimers are listed in Table 4.| Substituent | mp/°C | ||||
|---|---|---|---|---|---|
| X | Y | Monomer | ht Photodimer | hh Photodimer | Ref. |
| a By projection.b By hot stage microscope. | |||||
| CH3 | H | 80a | >250a | Calas and Lalande (1960)1,4 | |
| CN | H | 178a | ≈205a | Calas and Lalande (1960)1,4 | |
| F | H | 102a | 320a, 275b | Lapouyade28 | |
| COC6H5 | H | 148a | 214–218b | Becker1,2 | |
| OCH3 | H | 94a | 225–250b | 113–120b | Becker14 |
| CN | OAc | 199a | ≈150a | Calas and Lalande (1960)1,4 | |
The 9-cyano substituent induces an additional instability; moreover, when it is combined with the acetoxy substituent in the 9-cyano-10-acetoxyanthracene, the mp of the photodimer (probably of ht structure) is lower than that of the monomer (Table 4). It is also noticeable that the only isolated hh photodimer of a 9-substituted monomer has a lower mp than its ht isomer. It happens to parallel the relative thermal stability in solution.14
4.2 Molar mass determination
Early measurements were based on cryoscopy1 and ebulliometry (Calas and Lalande, 1959).1 Ebulliometry determination relies on the following known relationship linking the boiling point variation (ΔTbp) between a pure solvent and its diluted solution with the solute concentration: ΔTbp = KC/M where C is the solute mass concentration, K the solvent constant and M the solute molar mass. For example, the photoproduct isolated after irradiation of 9-vinylanthracene (410 mg in 78 g benzene) was found to display ΔT ∼ 0.043 in benzene (K = 2500) which gives an experimental value for M = 390. The theoretical mass for a dimer is 408. It was concluded that the product is a photodimer.9Mass spectrometry (electron impact at 70 V) generally fails to provide the molecular peak of the photodimer but displays the monomer molecular peak instead, often as the base peak (Chapman, 1969).1 However, Schmutzler reported the presence of the dimer signal at m/z 492 (0.1%) for the photodimer of 9-difluorophosphinoanthracene (Heuer et al., 1989).14 More recently, Becker successfully used the FAB(+) technique (with 3-nitrobenzyl alcohol as the matrix) to demonstrate the dimer structure of the hh 9,9′-dimethoxydianthracene.14
4.3 X-ray structure analysis
The method is sovereign but requires single crystals. It was used for dianthracene in 1932 by Hengstenberg and Palacios to assign the bonding positions and was refined by Ehrenberg (in 1966)1,4 and further by Choi et al. (1980).29 The C9-C9′ bond length was observed to be 162.4 pm. The same value has been found theoretically, using a B3LYP calculation employing a 6-31G* basis set.29 Further elongation was observed by Dunand et al. (1980)1 in related systems, e.g. for the C9–C9′ bond length (166.9 pm) of the photocycloisomer of 1,3-di(9-anthryl)propane (Scheme 14). The structures of other 9-substituted and 9,10-difluoroanthracene11 photodimers have also been established with the same remarkable lengthening of the bond linking the two halves. Substitution at the meso position also affects the dihedral angles A/B and A/A′ between the aromatic rings (Scheme 14). A result to be highlighted is the isolation and the structure determination of two conformational isomers of ht photodimer of 9-(2-hydroxy-2-propyl)anthracene14 (Scheme 14).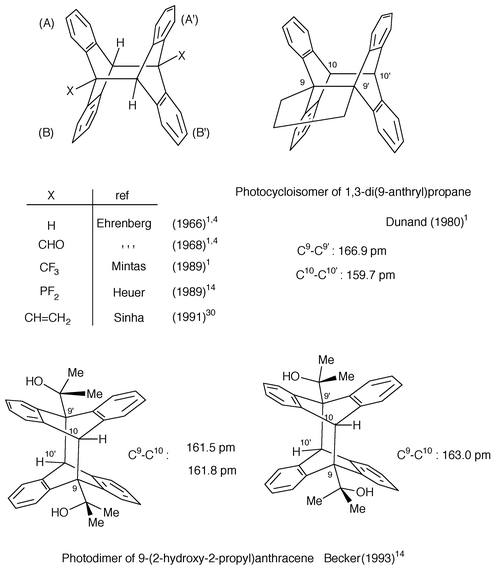 | ||
| Scheme 14 X-Ray molecular structure of some representative anthracene 9,10∶9′,10′ photocycloadducts. | ||
4.4 Dipole moment measurements
During the 1950–1960 period, one important issue was the structure determination of the 9-monosubstituted anthracene photodimers. As the skeleton geometry was known, it was anticipated that the permanent dipole moment (μ0) of the ht and hh isomers should be clearly different for polar substituents such as CN, Cl, Br. Experimental data were obtained by Calas et al. (1960)1 and Applequist et al. (1959)1 for the compounds listed in Table 5. For symmetrical substituents such as CN and Cl, the theoretical μ0 (ht) is expected to be zero. The experimental value would indeed reduce to zero if atomic polarization were to have been taken into account.These results together with those of chemical correlations led to the assumption that all photodimers (at least with polar substituents) had a ht structure.
4.5 Electronic absorption spectrometry
In involving the 9,10 (meso) positions, the photodimerization interrupts the conjugation of anthracene, generating four o-xylene chromophores. This was observed first by Coulson et al. (1955)1 who showed that ε270 nm and ε280 nm are equal to four times that of o-xylene (see Fig. 1). This result demonstrates the 9,10∶9′,10′ bonding. Moreover a considerable hypsochromic shift (≡8500 cm−1) is observed between the onsets of anthracene and 9,10∶9′,10′-dianthracene spectra. Fig. 1a illustrates the gradual disappearance of the 300–400 nm absorption band of an anthracene derivative and the growing of the 9,10∶9′,10′ classical photocyclomer during irradiation in the 300–400 nm range (Desvergne et al., 1995).24 Typical UV spectra are represented in Fig. 1b: the most frequent absorption found (curve i) is characteristic of the classical 9,10: 9′,10′ closure whereas curve (ii) indicates a naphthalene type absorption for 9,10∶1′,4′ photocycloaddition (Castellan et al., 1979);1 in a distinct way 9,10∶1′,2′ cycloaddition produces spectra type (iii) due to the presence of a vinyl-substituted naphthalene (Daney et al., 1985).1 Some other spectra should be considered e.g. of the 1,4-disubstituted xylene type (Fig. 1c) (Fages et al., 1988)16 or of the photodimers of 9-styrylanthracene (Fig. 1d) (Becker et al.).10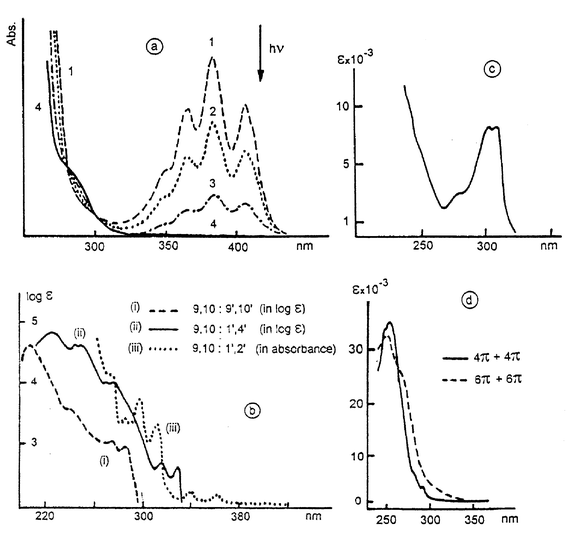 | ||
| Fig. 1 Some representative electronic absorption spectra of anthracene photodimers and photocycloisomers (with permission); (a) formation of a 9,10∶9′,10′ photodimer. Reprinted with the permission of Wiley-VCH. (b) Typical spectra of photocycloisomers (i) 9,10∶9′,10′; (ii) 9,10∶1′,4′; (iii) 9,10∶1′,2′ cycloadducts. (ii) Reprinted with the permission of CNRS. (iii) Reprinted from Tetrahedron Lett., 1985, 26, 1505, Copyright (1985), with permission from Elsevier Science. (c) 9,10∶9′,10′ photodimer of a 1,4-dialkoxyanthracene. (d) Photodimers of 9-phenylethynylanthracene (4π + 4π) and 9-styrylanthracene (6π + 6π). Reprinted with permission from J. Org. Chem., 1985, 50, 3913. Copyright (1985) American Chemical Society. | ||
4.6 NMR Spectrometry
The characterization of anthracene photodimers by NMR was made possible when high sensitivity equipment became available, i.e. in the early seventies. The 1H NMR chemical shifts of the tertiary bridgehead protons are characteristic of the structure whatever the type of cycloadduct: 9,10∶9′,10′, 9,10∶1′,4′ or 9,10∶1′,2′. Wilson et al.31 observed in 1969 a singlet at δ 4.56 in CDCl3 for the bridgehead protons of dianthracene after 500 scans. The same year, Chapman et al.1 demonstrated the ht structure of the 9-aminoanthracene photodimer: the authors obtained satisfactory 13C side-band spectra for the photodimer dissolved in formic acid using an accumulation technique; for a hh isomer the two 1H and 13C would form an AA′X spectrum. It was found JAA′ ≡ 0 and J13C–H 136 Hz; this result established the ht orientation for NH2. De Schryver et al. (1971)1 showed that 9-anthrylmethyl acetate leads to a 20∶80 mixture of hh∶ht isomers by irradiation in CH2Cl2 (Table 6). Kaupp et al. (1980)11 demonstrated the formation of a 40∶60 mixture of hh∶ht isomers of 9-methylanthracene photodimers; they assigned the signals disappearing by heating in refluxing benzene (δ 4.57) to the hh isomer. Most hh dimers were found to be more labile than the ht isomers in solution. Some selected data are listed in Table 6. Of special interest are the results of Wolff et al. (1983)1 who used 1H NMR to show the influence of micelles on the mutual orientation of the monomers: thus, for 9-hydroxymethylanthracene they could remarkably favour the hh over the ht isomers with SDS in water (Table 6); the polar substituent (CH2OH) was thus predominantly directed to the polar micelle water interface while the aromatic part was directed to the lipophilic part.| Substituent | hh | ht | % hh∶ht | Irradiation medium at RT | Ref. |
|---|---|---|---|---|---|
| a Wilson (1969), in ref. 1.b De Schryver (1971), in ref. 1.c Kaupp (1980), in ref. 7.d Desvergne (1981), in ref. 1.e Wolff (1983), in ref. 1.f Ref. 14.g Castellan (1979), in ref. 1.h Ref. 12. | |||||
| H | 4.56 | 4.56 | — | Toluene | a |
| –CH2OAc | 4.20 | 3.70 | 20∶80 | CH2Cl2 | b |
| –CH3 | 4.57 | 4.02 | 40∶60 | Benzene | c |
| –C6H5 | — | 5.57 | 0∶100 | Benzene | c |
| –CH2OCH3 | 4.67 | 3.73 | 40∶60 | Et2O | d |
| -CH2OH | 5.1 | 4.5 | 25∶75 | CH3OH | e |
| 60∶40 | H2O + SDS | e | |||
| (micelles) | |||||
| OCH3 | 4.49 | 4.41 | 45∶55 | Et2O | f |
| –SiMe3 | — | 4.02 | 0∶100 | Et2O | g |
| –CO-C6H5 | — | 6.18 | 0∶100 | Toluene | h |
| SDS = Sodium dodecyl sulfate. | |||||
Other data regarding the hh mixed photodimer between 9-methoxyanthracene and 9-cyanoanthracene (Castellan et al., 1975),1,14 hh and ht photodimers of 1,8-disubstituted anthracenes (Desvergne et al., 1978;16 Brotin et al., 19921) as well as some selected photocyclomers (Castellan et al., 19791) are given in Fig. 2. It is worthy of note that in all cases when the bridgehead protons form an AB spectrum, the coupling constant is ca. 10–11 Hz as expected from the rigid geometry revealed by X-ray analysis. Because of the effect of vicinal substituents and variations of the skeleton structure, most chemical shifts of the bridgehead protons have been found in the range δ 3.7–6.2 in CDCl3.
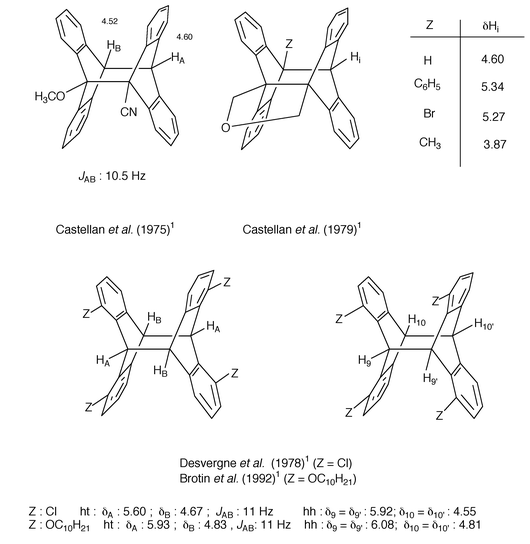 | ||
| Fig. 2 Chemical shifts (and coupling constants) of some representative bridgehead protons. | ||
4.7 Vibration spectrometry
Organic functions have characteristic infrared (IR) absorptions; some of them (e.g. the carbonyl group) are very sensitive to conjugation. 9-Anthraldehyde has a strong absorption at 1665 cm−1 which was found to move to 1723 cm−1 after photodimerization; this was taken as an indication that the CHO group was linked to a saturated carbon in the dimer, therefore strongly suggesting that the two anthracene halves (Greene, 1960)1 were bonded at the 9,9′ and 10,10′ positions.The presence of two clear absorption bands at ca. 1450 and 1470 cm−1 (splitting of C–H bonding) is a good indication of the presence of an anthracenic photodimer (De Schryver, 1971).1
In contrast, endoperoxides, which can be formed in aerated solvents in competition with photodimers when the photodimerization rate is very low, exhibit characteristic absorptions at 880–890 and 1230–1260 cm−1 (Nikitin et al.).32
5 Conclusion
The purpose of this article was to survey the structural aspects of anthracene photodimerization (intermolecular process) and photocycloisomerization (intramolecular process). Adopting a historical perspective may help in reading the abundant literature devoted to the subject. It appears that the field has considerably expanded over more than a century. The reactivity centres are no longer restricted to the central ring but may also involve the lateral rings and a variety of photodimer structures have been described. By means of NMR, UV spectrometry and, whenever possible, X-ray crystallography, the structure of the majority of photoproducts could be established but some failures to obtain photodimers or to isolate and characterize the photoproducts have been recorded.There are a number of synthetic methods available to allow the introduction of a variety of substituents in different positions, making it easy to incorporate one or several anthracene subunits in systems such as artificial membranes, polymers and other diverse materials where they can provide the functions of electron transfer relay, light emission switch, information storage. It is therefore very useful to know the photoreactivity of anthracenes and, particularly, their versatile cycloaddition ability.
The mechanistic schemes for this apparently simple reaction as well as the thermal and photochemical dissociation of the photodimers will be reviewed in a future article.
6 Acknowledgements
We are indebted to all our coworkers whose names are cited in the references. The CNRS, Université Bordeaux 1 and Région Aquitaine are warmly thanked for long term financial assistance. Finally we acknowledge NMR and Mass Spectra measurements by CESAMO. One of us (HBL) is particularly grateful to his mentor, the late Professor Calas, and to Professor Lalande for introducing him to the land of anthracenes.References
- H. D. Becker, Chem. Rev., 1993, 93, 145 and references therein. Search PubMed.
- D. O. Cowan and R. L. Drisko, Elements of Photochemistry, Plenum Press, New York, 1976, chap. 2. Search PubMed.
- H. D. Roth, Angew. Chem., Int. Ed. Engl., 1989, 28, 1193 CrossRef.
- H. Bouas-Laurent, A. Castellan and J.-P. Desvergne, Pure Appl. Chem., 1980, 52, 2633 and references therein. Search PubMed.
- J. Hengstenberg and J. Palacios, An. Soc. Esp. Fis. Quim., 1932, 5 Search PubMed.
- M. Ehrenberg, Acta Crystallogr., 1966, 20, 177; Acta Crystallogr., Sect. B, 1968, 24, 1123. Search PubMed.
- P. Suppan, Tetrahedron Lett., 1971, 4469 CrossRef CAS.
- R. Calas, R. Lalande, J.-G. Faugère and F. Moulines, Bull. Soc. Chim. Fr., 1965, 119 and 121. Search PubMed.
- R. Lalande, R. Calas and H. Bouas-Laurent, C. R. Séances Acad. Sci., 1959, 249, 706 Search PubMed.
- H. D. Becker, K. Andersson and K. Sandros, J. Org. Chem., 1985, 50, 3913 and references therein. Search PubMed.
- D. A. Dougherty, C. S. Choi, G. Kaupp, A. B. Buda, J. M. Rudzinski and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1986, 1063, and references therein. Search PubMed.
- H. D. Becker, V. Langer and H. C. Becker, J. Org. Chem., 1993, 58, 6394 and references therein. Search PubMed.
- R. Lapouyade, H. Bouas-Laurent and R. Calas, C. R. Séances Acad. Sci., 1968, 266, 1674 Search PubMed.
- H. D. Becker and V. Langer, J. Org. Chem., 1993, 58, 4703 and references therein. Search PubMed.
- H. Bouas-Laurent, R. Calas, M. L. Josien and R. Lalande, C. R. Séances Acad. Sci., 1960, 250, 4001 Search PubMed.
- H. D. Becker, H. C. Becker and V. Langer, J. Photochem. Photobiol. A. Chem., 1996, 97, 25 and references therein. Search PubMed.
- R. Lapouyade, J.-P. Desvergne and H. Bouas-Laurent, Bull. Soc. Chim. Fr., 1975, 2137, and references therein. Search PubMed.
- R. Lalande, R. Calas and H. Bouas-Laurent, C. R. Séances Acad. Sci., 1959, 249, 706 Search PubMed.
- A. Albini, E. Fasani and D. Faiardi, J. Org. Chem., 1987, 52, 155 CrossRef CAS.
- H. Bouas-Laurent and R. Lapouyade, C. R. Séances Acad. Sci., 1967, 264, 1061, and references therein. Search PubMed.
- R. Lapouyade, A. Castellan and H. Bouas-Laurent, C. R. Séances Acad. Sci., 1969, 268, 217 Search PubMed.
- J.-P. Desvergne, M. Gotta, J.-C. Soulignac, J. Lauret and H. Bouas-Laurent, Tetrahedron Lett., 1995, 36, 1259 CrossRef CAS.
- S. Lahlou, N. Bitit and J.-P. Desvergne, J. Chem. Res., 1998, (S) 302, (M) 1389. Search PubMed.
- J.-P. Desvergne, J. Lauret, H. Bouas-Laurent, P. Marsau, N. Lahrahar, H. Andrianatoandro and M. Cotrait, Recl. Trav. Chim. Pays-Bas, 1995, 114, 504 Search PubMed.
- F. Moriwaki, A. Ueno, T. Osa, F. Hamada and K. Murai, Chem. Lett., 1986, 1865 CAS.
- G. Deng, T. Sakaki, Y. Kawahara and S. Shinkai, Tetrahedron Lett., 1992, 33, 2163 CrossRef.
- J. Tucker, H. Bouas-Laurent, P. Marsau, S. W. Riley and J.-P. Desvergne, Chem. Commun., 1997, 1165 RSC.
- R. Lapouyade, H. Bouas-Laurent and R. Calas, C. R. Séances Acad. Sci., 1968, 266, 1674 Search PubMed.
- C. H. Choi and M. Kertesz, Chem. Commun., 1997, 2199, and references therein. Search PubMed.
- H. K. Sinha, A. J. Longh and K. Yates, J. Org. Chem., 1991, 56, 3727 CrossRef CAS.
- D. T. Wilson and B. K. Selinger, Photochem. Photobiol., 1969, 9, 171 Search PubMed.
- V. A. Nikitin and A. S. Cherkasov, Opt. Spektrosk., 1958, 4, 702 Search PubMed.
Footnote |
| † i.e. (1960, reviewed in refs. 1 and 4). Similarly throughout text. |
| This journal is © The Royal Society of Chemistry 2000 |