Electrochemistry of encapsulated redox centers
Claudia M. Cardona, Sandra Mendoza and Angel E. Kaifer*
Center for Supramolecular Science and Chemistry
Department, University of Miami, Coral Gables, FL 33124-0431, USA
First published on UnassignedUnassigned11th January 2000
Abstract
A few examples of molecules containing a redox active core surrounded by an organic sheath have been synthesized recently. The redox nucleus may be attached covalently to the structure’s core or held there by noncovalent forces. This review article surveys the electrochemical behavior of such redox centers, emphasizing the thermodynamic and kinetic effects brought about by encapsulation.
 Claudia M. Cardona Claudia M. Cardona | Claudia M. Cardona, born in Colombia, received her BS in chemistry from Brooklyn College of the City University of New York in 1984. After working for a cruise line company traveling the world for several years, she decided to continue her education and earned her MS in chemistry from Florida International University in 1994. She is currently completing her PhD at the University of Miami with support from a NIH-NIGMS Minority Predoctoral Fellowship. Her research has focused on the synthesis and characterization of electroactive and luminiscent dendrimers. |
 Sandra Mendoza Sandra Mendoza | Sandra Mendoza studied chemistry at the Universidad Nacional Autónoma de México, where she obtained her BS diploma in 1991 and her MS degree in 1993. She earned her PhD degree at the University of Miami in 1998 working on the synthesis and electrochemistry of hemicarcerands and their complexes. She is currently a faculty member at Universidad Autónoma de Querétaro, México. |
 Angel E. Kaifer Angel E. Kaifer | Angel E. Kaifer was born in Madrid, Spain, where he did his undergraduate work in chemistry at Universidad Autónoma. He earned his PhD in 1984 at the University of Puerto Rico, Rio Piedras Campus, and did postdoctoral work at the University of Texas, Austin. He joined the faculty of the University of Miami in 1985, where he is now Professor of Chemistry. His research interests focus on the electrochemistry of supramolecular systems. |
1 Introduction
The interplay between host–guest molecular recognition and redox chemistry has been the subject of two recent reviews.1,2 It is now clearly established that the stability of host–guest complexes can be strongly affected by changes in the oxidation state of the interacting partners. The literature presents numerous examples of these phenomena. We will initially focus our attention on the work of Evans and coworkers,3 originally published in 1985, concerning the binding interactions of a water-soluble ferrocene derivative (ferrocenecarboxylate) and the β-cyclodextrin (β-CD) host. These authors analyzed in detail the voltammetric behavior of ferrocenecarboxylate in the presence of β-CD. They observed that the presence of the host induces a shift of the apparent half-wave potential (E1/2) for the oxidation of ferrocenecarboxylate to more positive values. The half-wave potential is easily measured in voltammetric experiments and constitutes an excellent approximation to the formal potential (E°′) for the corresponding redox couple. The observed potential shift recorded by Evans and coworkers is consistent with the stabilization of the reduced form of the redox couple. They also observed that, in the presence of β-CD, the currents associated with the oxidation wave decrease. The latter effect is due to the marked decrease in the diffusion coefficient of the inclusion complex compared to that of the free guest (the CD host adds substantial bulk to the structure). These authors concluded that ferrocenecarboxylate and β-CD form a stable inclusion complex, while the oxidized, zwitterionic form of the guest does not interact strongly with the host. The oxidation of the guest in the presence of the host is best rationalized by an electron transfer step preceded by a coupled chemical reaction, i.e., the type of mechanism that electrochemists refer to as CE (chemical–electrochemical).The proposed mechanism is illustrated in Scheme 1. Notice that the mechanism does not include the direct oxidation of the inclusion complex. In fact, Evans and coworkers could not find any experimental evidence for direct electron transfer from the complex. At fast scan rates, the anodic voltammetric peak flattens, as the complex dissociation process becomes, relatively speaking, too slow to provide enough free ferrocenecarboxylate to sustain the fast-paced electrochemical oxidation. This is the best type of electrochemical evidence to support that no oxidation takes place on the β-CD inclusion complex.
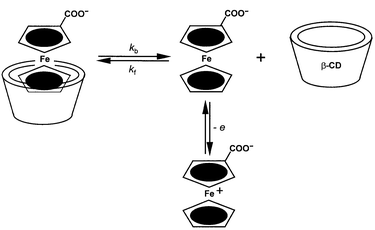 | ||
| Scheme 1 CE mechanism for the oxidation of ferrocenecarboxylate in the presence of the β-CD host. | ||
Over the years we have investigated a number of redox active guests in the presence of CD hosts.4–6 The results always resembled those found by Evans and coworkers. If an electroactive guest forms a stable inclusion complex with a CD host, the corresponding electrochemical reaction is suppressed in the complex. In all cases, complex dissociation must take place before the electron transfer step, as shown by the experimental evidence gathered while studying CD binding to several ferrocene derivatives,4 reduced viologens5 and cobaltocene.6 In order to understand these experimental results one must assume that encapsulation by CD hosts hinders the electron transfer processes of the guests, either thermodynamically, kinetically, or both.
The dynamic nature of these CD complexes leads to electron transfer from the equilibrating free guests. These findings opened interesting questions about the reactivity of encapsulated redox centers, defined as molecular fragments capable of electron transfer reactions that are fully surrounded by an organic sheath. How would encapsulation affect the kinetics and thermodynamics of electron transfer reactions if dissociation is prevented? This question should be extremely relevant to supramolecular chemists, as they routinely deal with systems in which molecular encapsulation plays a crucial role. Furthermore, this question is also of considerable relevance to those interested in biological electron transfer reactions since many redox active groups in proteins are partially or completely buried inside the protein’s polypeptide bulk. Only recently, progress in supramolecular chemistry has allowed us to look for experimental answers to these questions. This review will attempt to shed some light on these issues by surveying the electrochemical properties of two types of systems in which redox centers are encapsulated inside an organic sheath. These two types of systems are: (1) systems containing a redox center covalently attached to the core of a surrounding organic structure, and (2) systems containing a redox center maintained inside a hollow organic shell by noncovalent forces. The key difference between these two types of systems revolves around the mode of encapsulation (covalent or noncovalent) of the redox center.
2 Covalent encapsulation of redox centers: dendrimers with electroactive cores
The easiest way to accomplish the covalent encapsulation of a redox active center is to attach large organic branches (dendrons) to appropriate functional groups appended to the center of interest. Probably, the first reported work in this area was done by Diederich and coworkers, who prepared dendritic systems around a metal porphyrin core.7 The two dendrimers shown in Scheme 2 are representative structures of the two series of macromolecules synthesized by this group.8 Dendrimer 1 has a zinc porphyrin core and a hydrophobic surface that renders it soluble in nonpolar solvents, such as THF and CH2Cl2. In contrast, dendrimer 2 has an Fe core (with an additional chloride in the fifth coordination site) and more hydrophilic peripheral groups that make it soluble in water.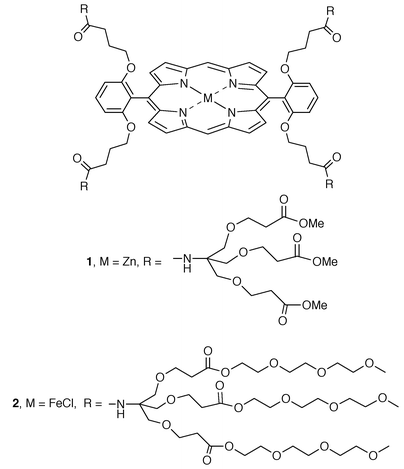 | ||
| Scheme 2 Diederich’s dendrimers containing a porphyrin core. | ||
The third generation analog of 1 has 108 methyl carboxylates on its surface and a molecular weight of 19054 daltons. Computer modeling suggests that it has a dense structure and globular shape, with a diameter of ca. 4 nm, a size similar to that of the redox protein cytochrome c. These dendrimers exhibit half-wave potentials for the first porphyrin-centered oxidation and reduction processes that shift to more negative values with increasing dendrimer generation.7,8 This finding is counterintuitive, and means that the generation of positive charge on the porphyrin residue is thermodynamically favored by the growth of the enveloping dendrimer mass. The electrochemical behavior of these dendrimers also reveals that the electron transfer reactions become slower as the dendrimer generation increases.
The series of iron dendrimers was also studied in aqueous solution.8 In going from dendrimer 2 to its next generation analog the potential for the Fe(III)/Fe(II) redox couple becomes more positive by 420 mV. This finding was rationalized by the increased hindrance experienced by water molecules as they solvate the more highly charged Fe(III) oxidation state through the increased dendrimer mass. When their electrochemical behavior was recorded in CH2Cl2, these same Fe dendrimers showed only a small difference in the half-wave potentials. These authors, in joint work with Collman’s group, have also reported on the O2 and CO binding affinity of the Fe(II) forms of these dendrimers9 (with imidazole replacing chloride at the fifth coordination spot). They found that the oxygen affinity is 1500 times higher than that of hemoglobin, while the CO affinity is similar. Both diatomic gases were bound reversibly by the Fe(II) porphyrin dendrimers. Aida and coworkers have also reported on the electron transfer reactions,10 and O2 and CO binding of Fréchet-type, porphyrin-containing dendrimers.11 Recently, Gorman and coworkers have described the synthesis and properties of a completely novel series of dendrimers12 containing a redox active, iron–sulfur core of the form [Fe4S4(SR)4]2−. This tetra-iron core exhibits a quasi-reversible one-electron reduction (Scheme 3). The voltammetric behavior of these dendrimers shows that the reduction processes become kinetically and thermodynamically hindered with increasing dendrimer generation (zeroth to fourth). The increased steric bulk imposed by the dendritic ligands R was interpreted to be responsible for the trends observed in the electrochemical data.
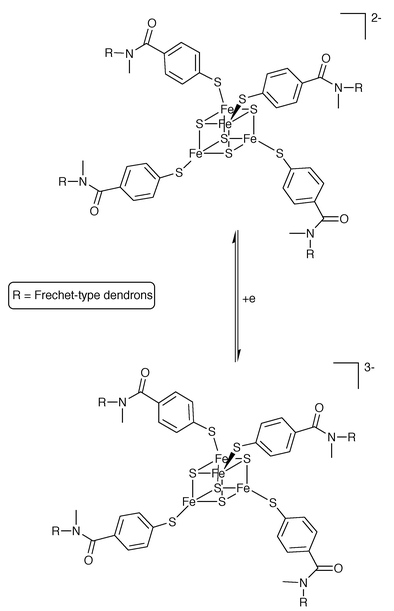 | ||
| Scheme 3 The electochemical reactivity of Gorman’s dendrimers. | ||
Newkome and coworkers have developed an interesting approach to the assembly of dendrimers that relies on the coordination of metal ions.13 The central idea is represented in Scheme 4. Two terpyridine-derivatized, dendritic wedges can be assembled into a larger structure by using an appropriate metal ion to which the terpyridines will act as ligands. These authors have used Ru(II) for the preparation of a series of dendrimers containing either a single13 or several14 bis(terpyridyl)Ru(II) centers. For the systems with a single metallic center, the kinetics of the electron transfer reactions were found to become slower as the macromolecular wedges become bulkier and wrap around the metal center.
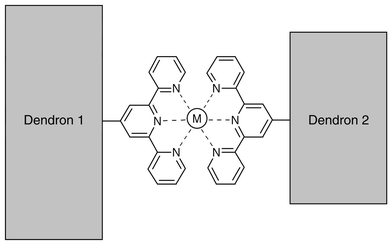 | ||
| Scheme 4 Newkome’s approach to dendrimer assembly through metal coordination. | ||
The group of Balzani and coworkers has also made extensive use of transition metal coordination in the design and preparation of many novel dendrimers.15 Their systems are synthesized fully relying on metal coordination, taking advantage of the directionality of ligand-to-metal bonds for the structural design of the dendrimers. The main building blocks used in this approach, along with Ru(II) and Os(II) nucleating metal ion centers, are shown in Scheme 5. The resulting dendrimers can be considered as organized assemblies of coordinated metal centers. In addition to very interesting spectroscopic properties, these dendrimers exhibit rich electrochemical behavior related to metal-centered one-electron oxidations or ligand-centered reductions. The magnitude of the interactions among the metal centers varies from small to considerable depending on their relative proximity. Therefore, the electrochemical behavior can be predicted if one considers these effects as well as others to result from the macromolecular nature of these systems. The utilization of different ligands and metal ions makes it indeed possible to design and establish electrochemical potential gradients in these dendrimers, an intriguing goal from the standpoint of electron and/or information storage applications at the molecular level.16
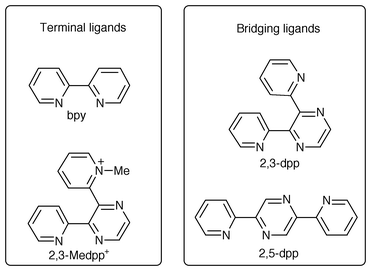 | ||
| Scheme 5 Set of ligands used by Balzani for dendrimer self-assembly. | ||
One feature that is commonly observed in redox proteins is the asymmetric positioning of the prosthetic group. Often, the redox active center is partially buried in the polypeptide backbone and located “off center” in the protein framework.17 This is one of Nature’s methods to control electron transfer reactivity in living systems, favoring reactions with desirable partners and hampering reactions with unwanted partners. Recently, the authors’ group begun to use dendrimer frameworks to mimic this type of directional redox reactivity. Using ferrocenecarboxylic acid as the redox active nucleus and Newkome’s synthetic methodology, we have synthesized a series of three novel dendrimers (compounds 3–5, Scheme 6) containing a single, asymmetrically located ferrocene center.18 The voltammetric behavior of these molecules was investigated in CH2Cl2 solution. As expected the rate constant for heterogeneous electron transfer decreased as the dendrimer generation increased. The half-wave potential for the one-electron oxidation of the ferrocene subunit shifts to less positive values in going from the first to the third generation compound. Again, this is not an intuitive finding, but is in excellent agreement with the trend observed by Diederich’s group on the oxidation of their dendrimers with Zn–porphyrin cores in nonpolar solvents7 (CH2Cl2 and THF). Hydrolysis of the peripheral esters in 3–5 produces a new series of three carboxylate-terminated, water-soluble dendrimers (compounds 6–8, Scheme 6). In pH 7 buffer solution, these dendrimers are almost fully ionized and exhibit a marked anionic character. Their voltammetric behavior resembles that of 3–5 in some aspects. For instance, the standard rate of heterogeneous electron transfer decreases with increasing dendrimer generation. However, the half-wave potential for the oxidation of the ferrocene subunit shifts to more positive values in going from compound 6 to 8, in contrast with the behavior observed with the hydrophobic analogs 3–5. In the water-soluble dendrimers, ease of hydration of the oxidized ferrocene moiety seems to be the predominant factor controlling the potential values. Our data suggest that in the second and third generation compounds (7 and 8) dendrimer growth obstructs the solvation of the ferrocenium center by water molecules.
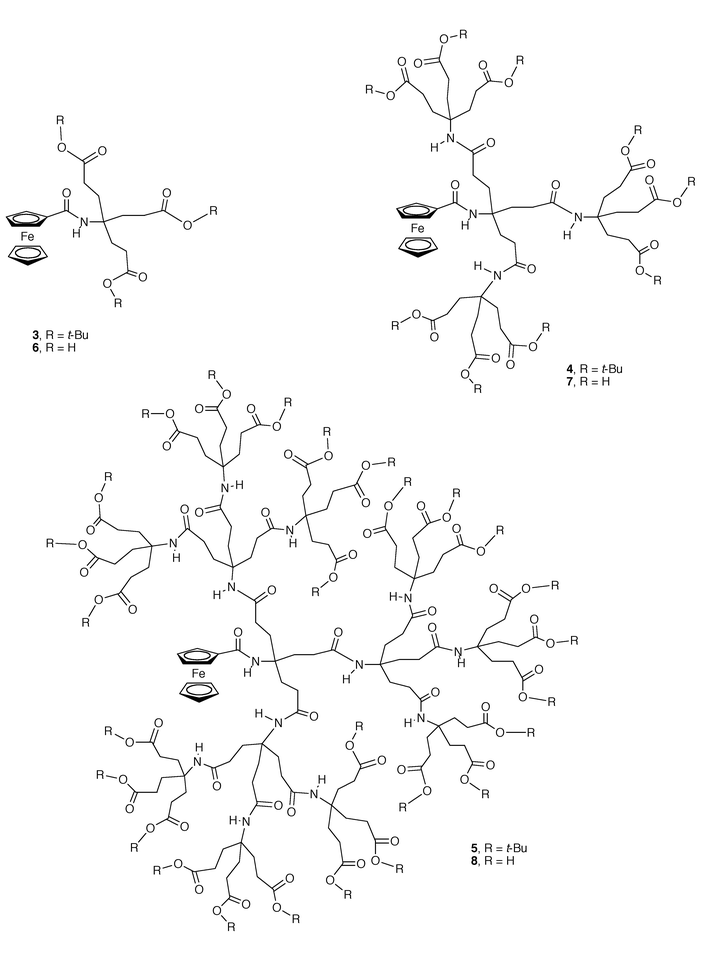 | ||
| Scheme 6 Unsymmetrical dendrimers containing a single ferrocene center. | ||
In summary, the electrochemical behavior of dendrimers with an electroactive core shows a clear common trend in all the systems that have been synthesized and investigated so far. Diffusion coefficients and rates of heterogeneous electron transfer decrease with increasing dendrimer generation. The variation of the diffusion coefficient is easy to rationalize as an increase in dendrimer generation is always accompanied by a substantial increase in molecular weight and effective radius. The slow down observed in electron transfer kinetics is probably related to the fact that the distance of maximum approach between the electroactive core and the electrode surface becomes larger as the dendrimer generation increases. Therefore, we can conclude that the covalent encapsulation of redox centers in dendrimer cores leads to kinetic hindrance of their electron transfer reactions. From a thermodynamic standpoint, the situation is more complex and encapsulation in a dendritic structure may shift the corresponding formal potential in either direction depending on the structure of the dendrimer and the medium in which the electrochemical experiments are carried out.
3 Noncovalent encapsulation of redox centers
Inclusion complexation is a form of molecular encapsulation, as the guest is surrounded partially or completely by the host in the complex. However, in order to investigate the electron transfer reactions of the encapsulated guest it is imperative to shut down or slow down host–guest dissociation and fully eliminate the free guest as a possible electron transfer partner. A possibility to do this would take advantage of interlocked molecules,19 such as catenanes and rotaxanes, to ‘lock’ redox active components in the inner regions of these topologically complex molecules. The groups of Stoddart and Balzani have investigated extensively the electrochemical behavior of numerous catenanes and rotaxanes containing redox active fragments.20 The main goal of their work has been to exert control on some structural and electronic features of interlocked molecules via electrochemical conversions, that is, to demonstrate their electrochemically switchable character. Some time ago, we also investigated in some detail the electrochemical behavior of rotaxanes inspired by Stoddart’s chemistry, but in these compounds the complete encapsulation of the redox center could not be ensured for all the accessible oxidation states.21 In general terms, the catenanes and rotaxanes already reported in the literature do not offer appropriate structural features for the effective encapsulation of redox fragments.Eventually, we turned our attention to molecular capsules, as a more effective approach for the noncovalent encapsulation of redox guests. The extensive work of Cram22 and more recently Sherman23 and Rebek24,25 has allowed supramolecular chemists to probe the properties of molecules inside hosts acting as capsules or containers. An interesting feature of these encapsulated molecules is that they are isolated from the solvent molecules, and can thus be considered to be in a ‘new phase of matter’. We should note here that the investigation of the electron transfer reactivity of encapsulated molecules is obviously an important aspect of their physical chemical characterization. As a first step in this direction, we selected hemicarcerands as optimum encapsulating hosts.22 These fascinating molecular receptors can form kinetically stable inclusion complexes (hemicarceplexes) with guests of appropriate size and shape. Crucial for the function of these hosts is a tight fit between the guest and the four equatorial portals (openings) in the structure of the hemicarcerand. The guest can only reach the hollow interior of the host by passing through one of the portals. If the size of the guest is close to the size of the opening, equilibration is exceedingly slow at room temperature. Inclusion complexation is faster at higher temperatures and, upon cooling, the guest is trapped inside the host because the dissociation process is also very slow. Cram termed this phenomenon constrictive binding.22 We reasoned that trapping of a redox active guest inside a hemicarcerand would allow us to investigate its electron transfer reactions without any need to consider the free guest or the dissociation process.
We prepared hosts 9 and 1026 (Scheme 7), which had been previously reported to encapsulate ferrocene by Quan and Cram.27 Preparation of the corresponding ferrocene inclusion complexes (Fc·9 and Fc·10) proceeded as described. While Fc·9 was only soluble in tetrachloroethane, Fc·10 was soluble in dichloromethane and tetrachloroethane. Guest release from Fc·10 in CD2Cl2 was monitored at 25 °C by 1H NMR spectroscopy. The half-life for the dissociation process was found to be long enough (t1/2 >300 h) to ensure that the electrochemical experiments could be done on the fully encapsulated ferrocene.
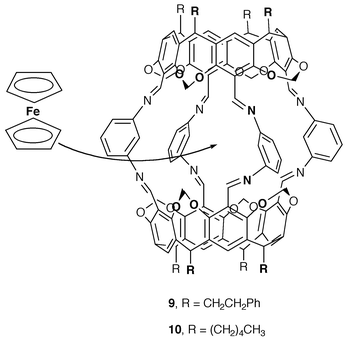 | ||
| Scheme 7 Ferrocene inclusion by hemicarcerands 9 and 10. | ||
Voltammetric experiments with Fc·9 in C2D2Cl4 and Fc·10 in C2D2Cl4 and CD2Cl2 yielded the following results26 in comparison to free ferrocene in the same media: (1) Encapsulation shifts the half-wave potential for ferrocene oxidation to more positive values. (2) Ferrocene encapsulation by either host causes a substantial decrease in both the diffusion coefficient and the standard rate of heterogeneous electron transfer.The first effect is probably related to the poor solvation that the inside walls of the hemicarcerands provide to the positively charged, oxidized ferrocenium guest. The second effect was interpreted using the same arguments utilized in the case of encapsulation in dendrimer cores. Therefore, hemicarcerand encapsulation of ferrocene hinders its heterogeneous electron transfer reactions both kinetically and thermodynamically.
We must note here that Deshayes and coworkers28 have investigated the triplet energy transfer between acetophenone encapsulated inside a related hemicarcerand and external acceptors. Their results suggest that the presence of the hemicarcerand shell causes only a modest decrease in the rate of triplet energy transfer, leading these authors to postulate an electron exchange mechanism involving the hemicarcerand skeleton. The groups of Pina and Balzani have also studied the effect of hemicarcerand inclusion on photophysical phenomena.29 For instance, these authors have investigated energy and electron transfer quenching processes of the lowest triplet excited state of biacetyl encapsulated in the same hemicarcerand host. Their results indicate that the walls of the hemicarcerand allow only a rather weak electronic interaction between the encapsulated triplet biacetyl and external quenchers. This work highlights that a lot more research work is necessary to understand the role of hemicarcerand hosts in electron and energy transfer processes through their walls.
4 Conclusions and outlook
In this review, we have tried to summarize the electrochemical behavior of systems that contain a redox center approximately surrounded by an organic sheath. The rapid development of the chemistry of dendrimers has afforded a number of examples in which a redox center is covalently attached to a dendrimer core and surrounded by its bulky structure. The complete noncovalent encapsulation of redox centers has been more unusual. To the best of the authors’ knowledge, the only reported example is the one described in this review. Generally, encapsulation tends to slow down the kinetics of heterogeneous electron transfer reactions. However, the chemistry of redox proteins suggests that systems in which the redox centers are positioned unsymmetrically may exhibit more exciting properties. For instance, unsymmetric redox encapsulation may lead to directional electron transfer properties30 and enhanced reactivity with selected partners. Research work addressing these issues is already underway and may lead to interesting and novel results.Acknowledgements
The authors are grateful to the NSF for the support of this research (to AEK, CHE-9633434 and CHE-9982014). CMC acknowledges support from NIH in the form of a minority predoctoral fellowship. AEK gratefully acknowledges the Iberdrola Visiting Scholars Program and Professor Moisés Morán who hosted him at Universidad Autónoma de Madrid while he completed this manuscript.References
- A. Niemz and V. M. Rotello, Acc. Chem. Res., 1999, 32, 44 CrossRef CAS.
- A. E. Kaifer, Acc. Chem. Res., 1999, 32, 62 CrossRef CAS.
- T. Matsue, D. H. Evans, T. Osa and N. Kobayashi, J. Am. Chem. Soc., 1985, 107, 3411 CrossRef CAS.
- R. Isnin, C. Salam and A. E. Kaifer, J. Org. Chem., 1991, 56, 35 CrossRef CAS.
- A. Mirzoian and A. E. Kaifer, Chem. Eur. J., 1997, 3, 1052 CrossRef CAS.
- Y. Wang, S. Mendoza and A. E. Kaifer, Inorg. Chem., 1998, 37, 317 CrossRef CAS.
- P. J. Dandliker, F. Diederich, M. Gross, C. B. Knobler, A. Louati and E. M. Sanford, Angew. Chem., Int. Ed., 1994, 33, 1739 CrossRef.
- P. J. Dandliker, F. Diederich, A. Zingg, J.-P. Gisselbrecht, M. Gross, A. Louati and E. Sanford, Helv. Chim. Acta, 1997, 80, 1773 CrossRef CAS.
- J. P. Collman, L. Fu, A. Zingg and F. Diederich, Chem. Commun., 1997, 193 RSC.
- R. Sadamoto, N. Tomioka and T. Aida, J. Am. Chem. Soc., 1996, 118, 3978 CrossRef CAS.
- D. L. Jiang and T. Aida, Chem. Commun., 1996, 1523 RSC.
- C. B. Gorman, B. L. Parkhurst, W. Y. Su and K.-Y. Chen, J. Am. Chem. Soc., 1997, 119, 1141 CrossRef CAS.
- G. R. Newkome, R. Güther, C. N. Moorefield, F. Cardullo, L. Echegoyen, E. Pérez-Cordero and H. Luftmann, Angew. Chem., Int. Ed., 1995, 34, 2023 CrossRef CAS.
- G. R. Newkome, F. Cardullo, E. C. Constable, C. N. Moorefield and W. C. Thompson, J. Chem. Soc., Chem. Commun., 1993, 925 RSC.
- V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Acc. Chem. Res., 1998, 31, 26 CrossRef CAS.
- For another example of a dendrimer system designed to exibit an electrochemical potential gradient, see: T. D. Selby and S. C. Blackstock, J. Am. Chem. Soc., 1998, 120, 12155.
- E. M. Bowden, Interface, 1997, 6(4), 40 Search PubMed.
- C. M. Cardona and A. E. Kaifer, J. Am. Chem. Soc., 1998, 120, 4023 CrossRef CAS.
- F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643 CrossRef CAS.
- V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405 CrossRef CAS.
- E. Córdova, R. A. Bissell and A. E. Kaifer, J. Org. Chem., 1995, 60, 1033 CrossRef CAS.
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests in Monographs in Supramolecular Chemistry, J. F. Stoddart, Ed., Royal Society of Chemistry, Cambridge, 1994, Vol. 4. Search PubMed.
- A. Jasat and J. C. Sherman, Chem. Rev., 1999, 99, 931 CrossRef CAS.
- J. Rebek, Acc. Chem. Res., 1999, 32, 278 CrossRef.
- M. M. Conn and J. Rebek, Chem. Rev., 1997, 97, 1647 CrossRef CAS.
- S. Mendoza, P. D. Davidov and A. E. Kaifer, Chem. Eur. J., 1998, 4, 864 CrossRef CAS.
- M. L. C. Quan and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2754 CrossRef CAS.
- A. Farrán, K. Deshayes, C. Metthews and I. Balanescu, J. Am. Chem. Soc., 1995, 117, 9614 CrossRef CAS.
- A. J. Parola, F. Pina, E. Ferreira, M. Maestri and V. Balzani, J. Am. Chem. Soc., 1996, 118, 11610 CrossRef.
- Y. Wang, C. M. Cardona and A. E. Kaifer, J. Am. Chem. Soc., 1999, 121, 9756 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |