Microscopic structure of nickel(II) co-ordination shell in NiCl2–methanol solution: neutron diffraction and ab initio studies
Oleg N. Kalugin† and Ashok K. Adya*
Division of Molecular and Life Sciences, School of Science and Engineering, University of Abertay Dundee, Bell Street, Dundee, UK DD1 1HG. E-mail: A.Adya@tay.ac.uk; Fax: ; Fax: +44 (0)1382 308663;; Tel: +44 (0)1382 308653
First published on UnassignedUnassigned22nd December 1999
Abstract
Neutron diffraction measurements were carried out on ∽1.4 molal solutions of NiCl2 in methanol under ambient conditions. First-, second- and high-order difference methods with isotopic substitution on nickel, chlorine and hydroxy hydrogen (HO) were applied in conjunction with ab initio calculations of Ni2+–methanol complex to derive Ni–O, Ni–HO, Ni–Cl, Ni–C and Ni–H(methyl) pair radial distribution functions, RDFs. Analyses of these RDFs demonstrates that Ni2+ is co-ordinated octahedrally by five methanol molecules and one chloride anion with the nearest-neighbour Ni–O, Ni–Cl and Ni–HO distances of 2.057(1), 2.348(2) and 2.619(6) Å, respectively. The results also reveal a preferential triangular orientation of the methanol molecules surrounding Ni2+ in its co-ordination shell.
I. Introduction
In spite of wide technical applications of non-aqueous electrolyte solutions (NAES),1 our ability to predict their thermodynamic and kinetic properties is still restricted by the simple ion–ion and ion–dipole electrostatic models. One reason for this is the absence of detailed information about solvation structure of ions, especially multicharged and transition metal cations. Such information essentially means a deeper understanding of these systems at a microscopic level.Among the various direct experimental methods (i.e., those methods for which the space and time dimensions are comparable to the molecular dimensions and characteristic time of molecular processes, respectively), neutron diffraction with isotopic substitution (NDIS) offers the most powerful technique for analysing the microscopic structure of ion–molecular systems.2,3 This method allows us to derive structural information of nearest-neighbour ion environment in terms of pair radial distribution functions (RDFs), interatomic distances and co-ordination numbers.
Solutions of NiCl2 in methanol are ideally suited for applying the NDIS technique due to (i) the availability of suitable nickel, chlorine and, hydrogen isotopes, and (ii) the high solubility of nickel chloride in this solvent. Among the non-aqueous solvents, methanol possesses interesting characteristics due to the presence of both hydrophobic (CH3) and hydrophilic (OH) groups. It forms a well established hydrogen bonding network which is responsible for many of the macroscopic properties of this solvent and its electrolyte solutions.4
Although solutions of NiCl2 in methanol have been investigated earlier by using the NDIS method,5 the results of this work have caused some controversy. Powell and Neilson5 reported that Ni2+ is co-ordinated to 3.7 methanol molecules and they estimated at least 0.8 chloride ions to be in contact with the nickel. The total co-ordination number, 3.7+0.8=4.5 is apparently inconsistent not only with the well established 6-coordinated octahedral structure of 3d- ransition metal(II) ions in aqueous solutions,6 but also with the results of nickel(II) co-ordination in non-aqueous oxygen-donating solvents7,8 without chloride as a counter ion. It made us wonder whether this behavioural change of Ni2+-coordination is caused with a change of the counter ion to Cl−. To answer this question, the pair RDF gNiCl(r) should be obtained experimentally and, as far as we know, this has not been reported so far in any non-aqueous solvent.
Further motivation for the present study has been provided by a necessity to elucidate spatial correlations of nickel with carbon and hydrogens of methanol's methyl group since this will allow us to obtain information about the orientation of methanol molecules around the cation in its co-ordination shell.
In this paper we present the results of a comprehensive structural study of Ni2+ co-ordination shell in a 1.4 molal solution of NiCl2 in deuterated methanol by using nickel, chlorine and hydroxy hydrogen isotopic substitution technique of neutron diffraction (ND). To further facilitate interpretation of experimental results at the pair RDF level, ab initio calculations on methanol molecule and [Ni(CH3OH)]2+ complex were also performed. The paper has the following structure. Section II describes the essential theory behind the NDIS method. The experimental method is given in Section III. The ND data analyses are described in Section IV with special attention being paid to the normalisation of scattering patterns. In Section V the results of NDIS experiments and ab initio calculations are presented and discussed in terms of Ni2+ pair RDFs, mean interatomic distances, co-ordination numbers and orientation of the methanol molecules around the nickel(II).
II. Theoretical background
The isotopic substitution technique of neutron diffraction, as applied to ionic solutions, has been described elsewhere.2 Only those relations necessary for understanding this article are described below.From a single neutron diffraction experiment on a multicomponent solution one obtains, after correction and normal isation, a differential cross-section (DCS), F0(k), which is given by the expression9
 | (1) |
where k=4π/λsin(θ) is the scattering vector defined by the scattering angle, 2θ , and the incident wavelength, λ; xi and σis are the atomic fraction and total bound (coherent and incoherent) scattering cross-section (TSCS) of species i, respectively, n is the total number of chemical species in the solution. The ε(k) is a term that arises from inelasticity effects owing to a departure from the static approximation.10,11 The total structure factor, F(k), is a linear combination of the partial structure factors
 | (2) |
where bi is mean coherent scattering length of species i. The partial structure factor, Sij(k), is related to the pair RDF, gij(r), by the Fourier transformation
 | (3) |
where na is the total atomic number density of the solution, averaged over all the species. It is worth noting that when i and j indices belong to the solvent atoms, both Sij(k) and gij(r) consist of two parts, intra- and intermolecular, often with overlapping contributions. The difference method of isotopic substitution enables cancellation of the intramolecular parts from the Sij(k) and gij(r) functions.
The Fourier transformation of the F(k) yields the total RDF, G(r), which is a linear combination of the pair RDFs, gij(r)

 | (4) |
where Wij=∑ij(2−δij)xixjbibj and δij is the Kronecker delta-function. For the solution of NiCl2 in methanol there are six (n=6) atomic species [Ni, Cl, O, HO(DO), C and H(D)], taking into account the different spatial positions of the hydrogens, H and HO of the methyl and hydroxy groups, respectively. Consequently, the number of different partial functions Sij(k) is equal to n(n+1)/2=21. However, hydrogen atoms of the methyl group are not all equal. While two methyl hydrogen atoms are closer to the hydroxy hydrogen and lie out of the symmetry plane of the methanol molecule, the third methyl hydrogen together with C, O and HO(DO) define this symmetry plane (see Fig. 1). The F(k) will then comprise of 28 partial structure factors, making it extremely difficult to interpret the total RDF, G(r) in terms of individual pair contributions.
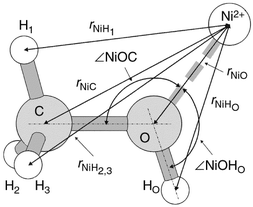 | ||
| Fig. 1 Sketch of the Ni(CH3OH)2+ complex optimised by MP2/TZV** ab initio calculations. | ||
In the first-order difference method, DCS is measured for two solutions denoted as m and l, identical except for the isotopic composition of one of the atomic species, αM and βM, which are characterised by different coherent scattering lengths, bαM and bβM, respectively. Difference in the two scattering functions, lF0(k) and mF0(k), gives
| lmΔF0(k)=lF0(k)−mF0(k) |

| =lmΔM1(k)+lmE′(k), | (5) |
where lmΔM1(k)=∑i=1nlmWMi[SMi(k)−1], with weighting factors

The real-space first-order difference function, lmGM(r), is obtained from

 | (6) |
An important property of the difference function lmΔM1(k) is that the ε(k) term in eqn. (1) will, to a first-order approximation, be identical for both the solutions.12 It cancels to give negligible contribution from the ε′(k) term in eqn. (5). The validity of this approximation can be checked on several accounts. Firstly, since gMi(r)=0 at r-values below the distance of closest approach of the M and i species (i.e., rmin), lmGM(r) should be equal to the calculated limit, lmGM(0)=−σlmWMi at low r. In practice, lmGM(r) will oscillate about this value owing to statistical noise on the k-space data, truncation of the data at a finite k-value and systematic errors. However, if these effects are small, setting lmGM(r) equal to the lmGM(0) limit for r⩽rmin and back Fourier transformation will yield the k-space first-order difference function in close agreement at all k-values with lmΔM1(k). Secondly, since Sij(k)→1 in the limit k→∞, lmΔF0(k) should oscillate about the calculated self-scattering difference, xM(σαMs−σβMs)/(4π).13 Thirdly, the k-space difference function should satisfy the sum-rule relation14
 | (7) |
In reality, it is difficult to apply the second and third criteria due to relatively short range of the scattering vector (k⩽20 Å−1) and significant inelasticity effects while using a reactor-based neutron source.
Additional isotopic substitution can be used to further separate the partial structure factors.15 The neutron second-order difference technique may be used to isolate the ion–ion and ion–hydrogen partial RDFs.2,3,15 Consider two first-order difference experiments identical except that apart from M, atom species γL is replaced by another isotope, δL. Assuming as usual that the structure of the solutions are not altered by the isotopic substitution, one obtains for the k- and r-spaces
| [lF0(k)−mF0(k)]−[pF0(k)−qF0(k)]=lmΔF0(k) |
 | (8) |
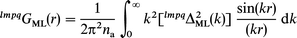
| =WML[gML(r)−1], | (9) |
where WML=2xMxL(bαM−bβM)(bγL−bδL).
For further consideration it is useful to introduce a co-ordination
number, ![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) Mi,
Mi,
 | (10) |
which defines the mean number of particles of type i contained in a volume restricted by two concentric spheres of radii r1 and r2, and centred on a particle of type M. If a peak in a real space difference function, gMi(r) or GM(r) may be associated with an individual pair, a co-ordination number can be calculated by integration of the corresponding function over the range of the relevant peak
 | (11) |
The superscript ‘eff’ indicates the possibility of multiple contributions to the peak.
III. Experimental
Sample solutions were prepared as follows. The solid mixtures of Zn37Cl2+K37Cl, used by Badyal and Howe16 in their previous experiments on molten salts and kindly sent to us, were converted into aqueous solution of H37Cl by using an Amberlit IR-120 (H) ion-exchange resin. To avoid any loss of expensive isotopes, the ion-exchange column was connected to a specially designed and calibrated conductometric sensor and a conductance bridge. The solid mixture of 62NiCl2+KCl also obtained from Howe,16 was converted into 62NiCO3·xH2O by reaction with an aqueous solution of Na2CO3 (Ultrapure, Alfa, Puratronic®, 99.995%) at ∽90°C. The light-green gelatinous precipitate of 62NiCO3·xH2O was thoroughly rinsed with deionised water by a combination of centrifuging and decantation processes, repeated until the centrifugate showed negative reaction of Cl−, Na+ and CO32− ions. Aqueous solutions of 62NiNCl2 , NNi37Cl2 and 62Ni37Cl2 were further prepared by mixing H*Cl and *NiCO3, where the asterisks refer to the isotopes. An aqueous solution of HNCl (BDH, Aristar®) was used for preparing 62Ni NCl2 salt. Powder of NNiCO3·2Ni(OH)2·4H2O (Alfa, Puratronic®, 99.996%) was used for preparing NNi37Cl2 salt. An aqueous solution of NNiNCl2 was obtained by dissolving crystalline NNiNCl2 (Alfa, Puratronic®, 99.995%) in a very dilute HNCl solution to avoid any hydrolysis. The solutions of *Ni*Cl2 were then evaporated to a solid form of *Ni*Cl2·6H2O and further dried on a vacuum line under 1–100 Pa by gradually increasing the temperature from 25 to 70°C during one week. Such a procedure reduced the water content to less then 0.01%, and this was estimated by frequent and accurate weighting of the samples. Anhydrous salts so obtained were yellow or yellow–brownish, as expected.17The solutions of NNiNCl2 and 62NiNCl2 in methanol-d3, CD3OH, (Cambridge Isotope Laboratories, 99.5% deuterated) and NNiNCl2, 62NiNCl2, NNi37Cl2 and 62Ni37Cl2 in methanol-d4, CD3OD (Cambridge Isotope Laboratories, 99.8% deuterated) were made by weight in a dried, nitrogen filled glove box. The solvents were preliminary dried under 4 Å molecular sieves during one week.
The sample densities were measured by using Anton Paar DMA 58 densimeter at 25.2°C, the average temperature during neutron diffraction experiments.
The neutron diffraction (ND) experiments were performed using the D4B instrument at the Institute Laue-Langevin, Grenoble, France. The incident wavelength was 0.7046 Å giving an available k-range of 0.2<k<17 Å−1. The scattered neutrons were detected with two 64-channel multidetectors, which were calibrated by using the scattering from a vanadium bar. Diffraction patterns were obtained for (i) the solutions in their containers, (ii) the empty containers, (iii) the instrumental background with the sample absent, (iv) a cadmium bar of 7 mm diameter in order to estimate the background correction at low scattering angles18 and, (v) a vanadium standard rod of diameter 6.08 mm. The two containers used were hollow vanadium cylinders of inner diameters 6.8 and 6.67 mm and outer diameters 7.02 and 7.053 mm, respectively. The cylinder's axis was oriented perpendicular to the incident beam of height 5.0 cm.
The details of the solutions used in the diffraction experiments are given in Table 1. The coherent scattering lengths of the 62Ni and 37Cl isotopes, as defined by mass spectrometry,16 were respectively, −6.75 and 3.68 fm (1 fm=10−15 m). The scattering lengths of the natural isotopes [C, 6.6460(12); O, 5.803(4); H, −3.7390(11); NNi, 10.3(1); NCl, 9.5770(8) fm] and deuterium [D, 6.671(4) fm] were taken from Sears.19 The scattering lengths, total bound and absorption cross-sections for deuterium in each solvent, CD3OH and CD3OD, were calculated by taking into account its enrichment in each one of them.
| Total scattering cross-section, σs/b | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Solute | Solvent | Designation of scattering pattern | Molality, m/mol kg−1 | Molar ratio, solvent to solute | Total atomic number density, na/atom Å−3 | Density, ρ/g cm−3 (at 25.2°C) | Bounda | Free atomb | Total absorption cross-sectioncσa/b |
| a Calculated using the bound scattering cross-sections for all the atoms.b Calculated using the free atom scattering cross-sections for methanol.c At a wavelength of 1.798 Å. | |||||||||
| NNiNCl2 | CD3OH | 1F0(k) | 1.395 | 20.5 | 0.0929 | 1.03864 | 19.26 | 3.05 | 0.624 |
| NNiNCl2 | CD3OD | 2F0(k) | 1.371 | 20.2 | 0.0924 | 1.05948 | 7.07 | 3.40 | 0.576 |
| 62NiNCl2 | CD3OH | 3F0(k) | 1.313 | 21.7 | 0.0927 | 1.03220 | 19.20 | — | 0.659 |
| 62NiNCl2 | CD3OD | 4F0(k) | 1.364 | 20.3 | 0.0924 | 1.06250 | 7.01 | — | 0.645 |
| NNi37Cl2 | CD3OD | 5F0(k) | 1.374 | 20.2 | 0.0924 | 1.06352 | 6.85 | — | 0.094 |
| 62Ni37Cl2 | CD3OD | 6F0(k) | 1.324 | 20.9 | 0.0922 | 1.06089 | 6.79 | — | 0.160 |
IV. Data analyses
The total differential cross-section, F0(k) can be obtained from the measured intensity, I(k), after applying various corrections in the form10| F0(k)=α(k)×[I(k)+δ(k)]/(4π), | (12) |
where α(k) accounts for normalisation, absorption and self-shielding and, δ(k) is the contribution from multiple scattering. Both the functions, α(k) and δ(k) depend on the total scattering and absorption cross-sections, σs and σa of the sample.
A major problem with normalisation of the DCS for NAES, unlike the case for aqueous solutions,20 is that the σs values are not known exactly and these must be estimated from some assumptions. There are two common practices to calculate TSCS by using either bound atom or free atom approaches,5,7 and there is no straightforward rule to choose either one of them. The consequence is that firstly, the σs values calculated by using the two approaches differ several times from each other and secondly, this leads to a significant difference not only in the differential cross-sections, but also in the RDFs, G(r) and the co-ordination numbers. In order to establish how and to what extent the variation in TSCS alters the experimental DCS, initial analyses of the data were made on the total scattering patterns of only two samples, ‘1’ and ‘2’ (see Table 1). The raw data for these solutions were corrected for scattering and absorption by the container, for self-absorption21 and for multiple scattering,22 and put on an absolute scale of b sr−1‡ by using scattering from the vanadium rod.23 All these corrections were performed by using three different values of the TSCS for each sample: 19.26, 6.0 and 2.0 b for NNiNCl2 in CD3OH and 7.08, 3.5 and 2.0 b for NNiNCl2 in CD3OD. These values cover the changes in σs from the bound to the free-atom TSCS values (see Table 1). The calculated F0(k) functions are shown in Fig. 2.
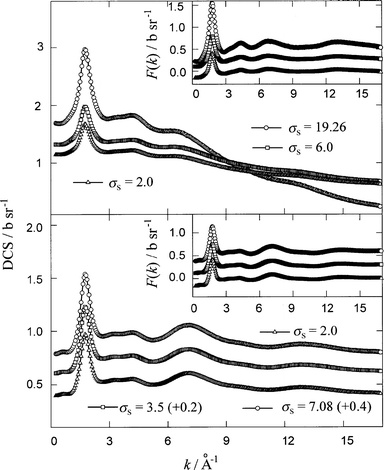 | ||
| Fig. 2 Experimental differential cross-sections, F0(k), for the solutions of NNi NCl2 in CD3OH (sample ‘ 1’, upper panel) and in CD3OD (sample ‘2’, lower panel) obtained by using different value, in b, of the total scattering cross-section (TSCS), σs, as indicated in the figure. Insets show the total structure factors, F(k), for the samples ‘1’ (upper panel) and ‘2’ (lower panel) after applying inelasticity correction to the F0(k) functions. For the inset in the upper panel, TSCS, σs=19.26 (+0.6) (circles), σs=6.0 (+0.3) (squares), σs=2.0 (triangles) and for that in the lower panel, σs=7.08 (+0.6) (circles); σs=3.5 (+0.3) (squares); σs=2.0 (triangles). | ||
Another problem with the evaluation of the total structure factor, F(k), is the inelasticity correction.24,25 The main effect of the inelasticity on hydrogenated and deuterated samples can be clearly seen in the droop of the DCS data at high k-values (see Fig. 2). In this work we have adopted an empirical approach based on the knowledge that the falloff is proportional to k2 in the low k-region,25 and at higher k-values there are additional terms which contribute to the Placzek droop. The total structure factors, F(k) were thus extracted from the F0(k) by subtraction of the polynomials, E(k) in the form of eqns. (13a) and (13b) for the samples ‘1’ and ‘2’, respectively.
| E3(k)=a+bk2+ck3+dk4, | (13a) |
| E4(k)=a+bk2+dk4. | (13b) |
The coefficients a, b, c and d of eqn. (13) were found by least-square fitting of E(k) to the experimental F0(k) functions. The F(k) functions, and the total RDFs, G(r) obtained by their Fourier transformation are shown in Figs. 2 (insets) and 3, respectively. For Fourier transformation we used our own program based on Numerical Recipes Fortran package.26 A comparison of the F(k) and G(r) functions for both the samples revealed that the use of different TSCS values for normalisation of the F0(k), followed by appropriate inelasticity correction, affects only the amplitude of these functions without any influence on their functional form. As an additional confirmation of this conclusion, one can see in Fig. 4 that the ratios G3.5(r)/G7.08(r), G2.0(r)/G7.08(r) and G2.0(r)/G3.5(r) for NNiNCl2 in CD3OD (sample ‘2’) are almost flat over the entire r-range.
 | ||
| Fig. 3 Total radial distribution functions, G(r), for the samples ‘1’ (upper panel) and ‘2’ (lower panel) obtained by Fourier transformation of the F(k) functions shown in Fig. 2 (insets). | ||
 | ||
| Fig. 4 The ratios of the total RDFs shown in Fig. 3 obtained by using different values of the total scattering cross-section for the sample ‘2’: G2.0(r)/G7.08(r) (circles), G3.5(r)/G7.08(r) (squares) and G2.0(r)/G3.5(r) (triangles). Full curves are the linear regressions over the data. | ||
It may be concluded from the above initial analyses that both the functions, F(k) and/or G(r) for a given sample can be easily renormalised by simply multiplying the corresponding functions calculated by using bound TSCS with an appropriate normalisation factor, nf, which is independent of the scattering vector, k, and/or interatomic distance, r.
As a most reliable criterion for the choice of the normalisation factor, the effective co-ordination number over the first peak in the total RDF, G(r) of NiCl2–methanol solutions can be used.5 This peak located at ∽1 Å in the total G(r) (see Fig. 3) is a weighted sum of the O–HO(DO) and C–D intramolecular correlations in accordance with the geometry of the methanol molecule (see Fig. 1 and Table 6 later). Thus, by integration of the total RDF one can obtain

 | (14) |
where
XO denotes either hydrogen or deuterium of the
hydroxy group, CD=3 and OXO=1 are the theoretical values of the co-ordination numbers for the D-atom around carbon
of the
methyl group and XO-atom around oxygen of the hydroxy group, respectively. Eqn. (14) yields the theoretical value
of effCD equal to 2.507 for CD3OH and 3.873 for CD3OD, respectively.
On the basis of the above discussion, the following scheme
of initial data analyses was adopted. (i) In the first stage, the
experimental scattering patterns for all the six solutions were
corrected for scattering and absorption by the container, for
self-absorption and for multiple scattering, and put on an absolute scale by using scattering from the vanadium rod. At this stage, only total bound (coherent plus incoherent) scattering cross-sections listed in Table 1 were used. (ii) The total structure factor, F(k), for each solution was then extracted from the observed DCS function, F0(k). For applying the inelasticity corrections, polynomials in the form of eqn. (13), E3(k) for the solutions in methanol-d3 and E4(k) for the solutions
in methanol-d4, were used. For each sample, the coefficients
a, b, c and d were treated as adjustable parameters in the
least squares refinement. It should be noted (see eqn. (1)) that the polynomials E3(k) and E4(k) incorporate not only the inelasticity effects, but also the self scattering term, σxiσis/(4π) of the measured DCSs. (iii) In the final step of the initial analysis procedure, the normalisation factors, nf3 and nf4, for the solutions in CD3OH and CD3OD, respectively, were found by scaling (see eqn. (14)) the total RDFs, G(r), obtained by Fourier transformation of the corresponding F(k) functions. This
operation was continued until the effective co-ordination numbers, effCD, calculated by integration of the scaled total
RDFs, G†(r)=nfi×G(r) (see Fig. 5) over the first peak yielded their theoretically expected values. It was found (see Table 2) that the normalisation factors should be nf3=0.466 and nf4=0.747 to satisfy the above described criterion. Using these values of nf3 and nf4, the experimental DCS functions were renormalised according to the relations
 | ||
| Fig. 5 Renormalised total RDFs (scaled for intramolecular C–D and O–XO (X=H or D) correlations) for the solutions of nickel chloride in methanol-d3 (upper panel) and methanol-d4 (lower panel): 1,2—NNiNCl2, 3,4—62NiNCl2, 5—NNi37Cl2 and 6—62Ni37Cl2. | ||
CD calculated by integration of the renormalised total RDFs, G†(r)=nfi×G(r) (Fig. 5) over the first peak, for 1.4 molal solutions of NiCl2 in methanol
| Sample | effCD | Integration range/Å |
|---|---|---|
| a The theoretical value is 2.507 for solutions of *Ni*Cl2 in CD3OH.b The theoretical value is 3.873 for solutions of *Ni*Cl2 in CD3OD. | ||
| CD3OH (nf3=0.466) | ||
| 1 | 2.52 | 0.9–1.3 |
| 3 | 2.48 | 0.9–1.3 |
| Mean value | 2.50a | |
| CD3OD (nf4=0.747) | ||
| 2 | 3.85 | 0.825–1.325 |
| 4 | 3.93 | 0.825–1.325 |
| 5 | 3.84 | 0.825–1.325 |
| 6 | 3.89 | 0.825–1.325 |
| Mean value | 3.88b | |
| F0†(k)=nf3×F0(k) | (15a) |
and
| F0†(k)=nf4×F0(k) | (15b) |
for the solutions of *Ni*Cl2 in CD3OH and CD3OD, respectively. The renormalised F0†(k) functions, shown in Fig. 6, were used for further analyses as described in Section V. In the sub sequent discussion we omit the ‘†’ sign as a superscript in the DCS functions bearing in mind that these functions have already been renormalised.
 | ||
| Fig. 6 Renormalised experimental differential cross-sections for the ∽1.4 molal solutions of nickel chloride in methanol-d3 (upper panel) and methanol-d4 (lower panel): 1,2—NNiNCl2, 3,4—62NiNCl2, 5—NNi37Cl2 and 6—62Ni37Cl2. | ||
V. Results and discussion
A. First-order differences
The three first-order difference functions, 13ΔNi1(k), 24ΔNi1(k) and 56ΔNi1(k), describing the co-ordination structure around the nickel cation are illustrated in Fig. 7. These were obtained from the renormalised DCSs, F0(k)s, (see Section IV) for the pairs of samples 1 and 3, 2 and 4, and 5 and 6 (for designation of the samples, see Table 1), respectively. To account for the residual self-scattering and inelasticity effects (eqn. (5)), polynomials of the form | ||
| Fig. 7 First-order difference functions, lmΔNi1(k), where l and m are the designations (Table 1) of the two scattering patterns used for their evaluation. Symbols are the data points and curves are the back Fourier transforms of the real-space lmGNi(r) functions after setting them to their low r-value limits, lmGNi(0) (full curves in Fig. 8). | ||
 | ||
| Fig. 8 First-order real-space difference functions, lmGNi(r) vs. r. The dotted curves are the Fourier transforms of the measured lmΔNi1(k) functions of Fig. 7 and full curves are lmGNi(r) after the unphysical oscillations at r⩽1.85 Å have been set to their low r-value limits, lmGNi(0) (see Table 3). | ||
| lmE′(k)=a+bk2+ck4 | (16) |
were subtracted from each difference function lmF0(k). The coefficients a, b and c for each of the difference functions were obtained by least-square fitting procedure.
The three real-space difference functions, 13GNi(r), 24GNi(r) and 56GNi(r), obtained by Fourier transformation of lmΔNi1(k), are shown in Fig. 8. The unphysical features at r⩽1.85 Å oscillate about the correct lmGNi(0) values (see Table 3). Also, the back Fourier transforms of lmGNi(r) obtained after setting lmGNi(r)=lmGNi(0) at r⩽1.85 Å (see Fig. 7) are in good agreement with the corresponding lmΔNi1(k) functions from which they are obtained. This demonstrates that the residual inelasticity corrections incorporated by the lmE′(k) polynomials (eqn. (16)) are small, that the cross-sections used in data analyses are reasonable, and that the obtained real-space first-order difference functions are correctly behaved.
The RDFs shown in Fig. 8 are linear combinations of the weighted nickel(II) pair distribution functions
| Difference (lm) | |||
|---|---|---|---|
| Correlation | 1–3 | 2–4 | 5–6 |
| Ni–Ni | 0.042 | 0.040 | 0.041 |
| Ni–Cl | 0.395 | 0.421 | 0.158 |
| Ni–O | 2.503 | 2.583 | 2.557 |
| Ni–HO(DO) | −1.613 | 2.960 | 2.931 |
| Ni–C | 2.867 | 2.959 | 2.930 |
| Ni–D | 8.565 | 8.881 | 8.792 |
| lmGNi(0) | 12.759 | 17.845 | 17.409 |
| lmGNiX*(0) | 13.977 | 14.462 | 14.320 |
 | (17) |
As the values of the weighting factors, lmWNiX (Table 3) suggest, the difference functions lmGNi(r) are mainly dominated by contributions from the correlations of Ni2+ with methanol atoms, but the influence of Ni–Cl correlation cannot be ignored. It should be noted (eqn. (17)), that the two kinds of methyl hydrogens, D1(H1) and D2,3(H2,3) are made distinguishable according to their different spacial orientation with respect to the hydroxy group and symmetry plane of the methanol molecule (Fig. 1).
All the three first-order difference functions, 13GNi(r), 24GNi(r) and 56GNi(r) reveal four sharp and well-separated peaks (see Fig. 8). To obtain reliable quantitative characteristics of these peaks, each lmGNi(r)−lmGNi(0) function was fitted by a sum of four Gaussians given by the expression
 | (18) |
where A, σ and rmax are, respectively, the area, half-width at half-maximum and mean position of the peak. The results of the fitting are shown in Fig. 9, and the fitted parameters are listed in Table 4. The first peak occurring at 2.065–2.097 Å can obviously be assigned to Ni–O nearest-neighbour correlations on the basis that the methanol molecule acts as an oxygen-donating ligand. The second peak at 2.582–2.729 Å is negative for the NiCl2 solutions in CD3OH and positive for those in CD3OD. This peak, thus, corresponds to Ni–HO (hydroxy hydrogen) correlations since the coherent scattering length of hydrogen is negative. The third and fourth peaks are then readily associated with the correlations of the nickel cation with the carbon and hydrogen (deuterium) of the methyl group, although exact assignment of these peaks to individual correlations can not be made from the present NDIS experiments. To clarify the spatial disposition of the methanol molecule with respect to Ni2+, ab initio calculations were performed (see Section V.C).
![[lmGNi(r)−lmGNi(0)] functions fitted by a sum of four Gaussians represented by the functions fi(r) of eqn. (18). The symbols represent the data, the dotted curves give the individual fitted Gaussians and the full curve gives their sum.](/image/article/2000/CP/a907829k/a907829k-f9.gif) | ||
| Fig. 9 [lmGNi(r)−lmGNi(0)] functions fitted by a sum of four Gaussians represented by the functions fi(r) of eqn. (18). The symbols represent the data, the dotted curves give the individual fitted Gaussians and the full curve gives their sum. | ||
| Difference (lm) | |||
|---|---|---|---|
| Peak and its parameters | 1–3 | 2–4 | 5–6 |
| a rmax, σ and A are the mean position, half-width at half-maximum and area of a peak. effNiO and effNiHO are the effective co-ordination numbers of the oxygen and hydroxy hydrogen around the Ni2+ obtained by integration of the corresponding Gaussians over the ranges ΔrNiO and ΔrNiHO, respectively. | |||
| 1st peak | |||
| rmax/Å | 2.065(1) | 2.097(1) | 2.082(1) |
| σ/Å | 0.115 | 0.117 | 0.109 |
| A/b | 0.0162 | 0.0170 | 0.0162 |
| effNiO | 5.3(3) | 5.5(3) | 5.2(3) |
| ΔrNiO/Å | 1.65–2.45 | 1.7–2.5 | 1.7–2.5 |
| 2nd peak | |||
| rmax/Å | 2.729(9) | 2.581(2) | 2.600(3) |
| σ/Å | 0.127 | 0.147 | 0.151 |
| A/b | −0.0043 | 0.0137 | 0.0135 |
| effNiHO | 3.8(5) | 5.9(4) | 5.9(4) |
| ΔrNiHo/Å | 2.2–3.2 | 2.1–3.1 | 2.1–3.1 |
| 3rd peak | |||
| rmax/Å | 3.163(4) | 3.158(2) | 3.160(3) |
| σ/Å | 0.179 | 0.182 | 0.167 |
| A/b | 0.0167 | 0.0196 | 0.0181 |
| 4th peak | |||
| rmax/Å | 4.007(7) | 4.012(6) | 4.002(9) |
| σ/Å | 0.189 | 0.173 | 0.191 |
| A/b | 0.0098 | 0.0104 | 0.0107 |
Table 4 reveals that there are certain discrepancies in the positions of the first and second peaks obtained from the three first-order difference functions. Although the positions of the first peaks are in good agreement with those reported by Powell and Neilson,5 the three values of 2.065, 2.097 and 2.082 Å are slightly greater than the Ni–O distance of 2.05 Å derived by Inada et al.8 from a recent EXAFS experiment on 0.3 molal solution of NiCl2 in methanol. Moreover, the position of the second peak in the case of 13GNi(r) RDF is shifted by ∽0.1 Å towards high r as compared to those for 24GNi(r) and 56GNi(r) RDFs (see Table 4).
The effective co-ordination numbers of the oxygen and hydrogen of the hydroxy group around the nickel, obtained by integration of the first two normalised Gaussians
 | (19) |
 | (20) |
are
also reported in Table 4. As it follows from the molecular
structure of the methanol molecule, the co-ordination numbers effNiO and effNiHO, for a given lmGNi(r) RDF, should be equal to each other. Table 4, however, shows that these values are different, the difference being −1.5, 0.4 and 0.7 for 1–3, 2–4
and 5–6 difference, respectively.
The above discrepancies in both the peak positions and the co-ordination numbers, as will be shown in the following sections, can be easily explained by considering the penetration of the first solvation shell of Ni2+ by chloride ion.
B. The pair RDFs, gNiHO(r) and gNiCl(r)
On the assumption that co-ordination structure around Ni2+ is unaffected by any isotopic substitution, e.g., NNi by 62Ni, HO by DO, and NCl by 37Cl, the two second-order difference functions, 2413ΔNiHO2(k) and 2456ΔNiCl2(k) were evaluated (Fig. 10) from the corresponding first-order difference functions | ||
| Fig. 10 Second-order difference functions, lmpqΔNiX2(k) for the Ni–Cl (upper panel) and Ni–HO (hydroxy hydrogen) (lower panel) correlations. Symbols are the data points and curves are the back Fourier transforms of the real-space lmpqGNiX(r) functions after setting them to their low r-value limits, lmpqGNiX(0) (full curves in Fig. 11). The statistical noise (errors) cannot be seen for 2413ΔNiHO2(k) on the given scale. | ||
| 2413ΔNiHO2(k)=24ΔNi1(k)−13ΔNi1(k) |
| =2413WNiHO[SNiHO(k)−1], | (21) |
| 2456ΔNiCl2(k)=24ΔNi1(k)−56ΔNi1(k) |
| =2456WNiCl[SNiCl(k)−1] | (22) |
without applying any additional inelasticity correction. Their Fourier transformations yielded the real-space functions, 2413GNiHO(r) and 2456GNiCl(r), which are shown in Fig. 11. The back Fourier transforms of these functions, after the unphysical oscillations at r⩽rmin have been set to the calculated values of lmpqGNiX(0)=−lmpqWNiX (X=HO, Cl) (Table 5), are found to be in good agreement with the original k-space functions (see Fig. 10). This validates the correctness of the procedure used for determination of the second-order difference functions. The pair RDFs, gNiHO(r) and gNiCl(r) as given by
 | ||
| Fig. 11 Second-order real-space difference functions, 2456GNiCl(r) (upper panel) and 2413GNiHO(r) (lower panel) vs. r. The dotted curves are the Fourier transforms of the measured lmpqΔNiX2(k) functions of Fig. 10 and full curves are lmpqGNiX(r) after the unphysical oscillations at r⩽rmin have been set to their low r-value limits, lmpqGNiX(0). | ||
| Correlation | ||
|---|---|---|
| Parameter | Ni–Cl | Ni–HO |
| a rmax, σ and A are the mean position, half-width at half-maximum and area of a Gaussian. NiX and ΔrNiX are the corresponding co-ordination numbers and their integration ranges. | ||
| lmpqWNiX/mb | 0.262 | 4.573 |
| rmax/Å | 2.348(2) | 2.619(6) |
| σ/Å | 0.104 | 0.189 |
| A/G | 7.1 | 4.4 |
| NiX | 0.72(5) | 5.4(5) |
| ΔrNiX/Å | 2.0–2.7 | 2.0–3.2 |
 | ||
| Fig. 12 The pair RDFs, gNiCl(r) (upper panel) and gNiHO(r) (lower panel). The symbols correspond to the data and the full curves give the fitted Gaussians represented by the function fi(r) of eqn. (18). | ||
| gNiHO(r)=2413GNiHO(r)/2413WNiHO+1, | (23) |
| gNiCl(r)=2456GNiCl(r)/2456WNiCl+1 | (24) |
along
with the fitted individual Gaussian functions in the form
of eqn. (18) are shown in Fig. 12. The object of the Gaussian
fitting of individual RDFs is to increase the accuracy in peak
positions and co-ordination numbers, especially in the case of gNiHO(r) where small artificial shoulders apart from the main peak are also observed (Fig. 12). The fitted and other important
parameters of the second-order difference functions are summarised in Table 5. It can be seen that in 1.4 molal solution
of NiCl2 in methanol, the mean Ni–HO distance at 2.619(6) Å is comparable to that in aqueous20 (2.67–2.80(2) Å) and ethylene glycol7 (2.82–2.86(2) Å) solutions with the same
concentration of Ni2+. The Ni–Cl nearest-neighbour distance of 2.348(2) Å is very close to the sum of the Pauling ionic radii,27 2.41=0.6+1.81 Å. This is an obvious evidence of direct cation–anion complexation (‘contact ion pair’1) in contrast to the results for aqueous NiCl2 solutions.28 A compari
son of the positions of the first and second peaks in the real-space first-order difference functions (Table 4, Fig. 9) with the average Ni–Cl distance (Table
6) indicates that nickel–chlorine correlation contributes significantly to the first and second peaks in lmGNi(r), and also to the effNiO and effNiHO co-ordination numbers.
| Methanol molecule | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ab initio calculations | Experiment | Ni2+–methanol complex Ab initio calculations | ||||||
| Parameter | MP2/3-21G | MP2/TZV** | Gas phaseb | Liquid phasec | MP2/3-21G | MP2/TZV** | ||
| a 1 Eh=627.51 kcal mol−1.b Ref. 30.c Ref. 31. | ||||||||
| rOHO | 0.992 | 0.960 | 0.9451 | 0.961 | 0.997 | 0.978 | ||
| rCO | 1.471 | 1.421 | 1.4246 | 1.415 | 1.577 | 1.541 | ||
| rCH1 | 1.090 | 1.085 | 1.0936 | 1.096 | 1.090 | 1.086 | ||
| rCH2,3 | 1.098 | 1.091 | 1.0936 | 1.096 | 1.088 | 1.083 | ||
| ∠COHO | 107.0 | 107.9 | 108.53 | 111.0 | 108.3 | 113.1 | ||
| ∠OCH1 | 105.5 | 106.6 | — | — | 105.8 | 105.6 | ||
| ∠OCH2,3 | 111.2 | 112.1 | — | — | 106.1 | 105.4 | ||
| rNiO | — | — | — | — | 1.775 | 1.752 | ||
| rNiHO | — | — | — | — | 2.479 | 2.407 | ||
| rNiC | — | — | — | — | 3.004 | 2.936 | ||
| rNiH1 | — | — | — | — | 2.971 | 2.887 | ||
| rNiH23 | — | — | — | — | 3.642 | 3.565 | ||
| ∠NiOC | — | — | — | — | 127.2 | 125.9 | ||
| ∠NiOHO | — | — | — | — | 124.4 | 121.0 | ||
| E | −114.6093 | −115.5188 | — | — | −1613.5240 | −1621.7095 | ||
Integration of the fitted gNiHO(r) and gNiCl(r) RDFs over the
ranges 2.0⩽r⩽3.2 Å and 2.0⩽r⩽2.7 Å, respectively, gives NiCl=0.72(5) and NiHO=5.4(5) (Table 5). This finding is consistent
with the 6-site octahedral co-ordination of Ni2+ that corresponds
to the sp3d2 hybridisation of its atomic orbitals.6
This type of co-ordination is typical for nickel cation in its
aqueous6 and non-aqueous7,8 solutions. From this point of view, the value of 3.7(3) for the NiHO co-ordination number obtained
by Powell and Neilson5 for 1 molal NiCl2–methanol solution seems to be underestimated.
C. Ab initio calculations
Ab initio calculations were performed by using the GAMESS program29 on Digital Alpha Workstation at the University of Abertay Dundee, Dundee, Scotland, UK. The two basis sets, 3-21G and TZV** (triple zeta with polarisation functions) of different quality were used for a full geometry optimisation of the methanol molecule and Ni2+–methanol complex. The structures optimised at the restricted Hartree-Fock (RHF) level were further optimised at the second-order Møller–Plesset perturbation (MP2) level. The optimised structural parameters of the methanol molecule and Ni(CH3OH)2+ complex along with total molecular energies are presented in Table 6. The equilibrium geometry of the Ni(CH3OH)2+ complex is shown diagrammatically in Fig. 1. The intramolecular geometrical parameters of the methanol molecule obtained experimentally in gas30 and liquid phases31 are also included in Table 6.The results of our calculations can be summarised as follows. Despite the fact that the total molecular energy of CH3OH obtained at the MP2/TZV** level of theory differs significantly from the corresponding 3-21G value, the struc tural parameters of an isolated methanol molecule calculated by using both the basis sets are in reasonable agreement with the experimental results.
The co-ordination of methanol molecule with Ni2+ alters significantly the C–O bond length and the ∠OCH2(3) for both the basis sets (see Table 6). The ∠COHO , calculated at the MP2/TZV** level, is also changed upon co-ordination. The C–O bond length increases by ∽0.11 Å and the ∠OCH2(3) decreases by 5–6° whereas the ∠COHO obtained at MP2/TZV** level increases by ∽5°. As shown in Table 6, the O–HO and C–H bond lengths are almost insensitive to the co-ordination of methanol with Ni2+.
The most interesting result in Table 6 is that the interatomic Ni–C and Ni–H1 distances obtained at the same level of theory coincide with each other within 0.03–0.06 Å. However, they differ significantly from the Ni–H2(3) distance. Upon the basis of the results given in Table 6, the third and fourth peaks in the RDFs, lmGNi(r) obtained by using the first-order difference method of ND (see Fig. 8 and 9) can be safely assigned. Thus, the third peak corresponds to a combination of the Ni–C and Ni–H1(D1) interatomic correlations seen at ∽3.16 Å, whereas the fourth one occurring at ∽4 Å results from overlapping Ni–H2(D2) and Ni–H3(D3) correlations. The difference of 0.2–0.4 Å in the positions of the fourth peak in lmGNi(r) (Table 4) and Ni–H2(3) interatomic distances as calculated by ab initio method (Table 6) can be associated with the absence of other co-ordinating species, such as Cl− and four additional methanol molecules in the modelled Ni2+–methanol complex.
D. High-order differences
Using the partial structure factors SNiHO(k) and SNiCl(k), obtained by using the second-order difference technique of ND (see Section V.B), the high-order k-space difference functions | (25) |
where X=(Ni, O, C, D1–D3) and lm=(13, 24, 56), were also extracted (Fig. 13) at the level of accuracy relevant to this work. The corresponding RDFs, lmGNiX*(r) obtained by Fourier transformation of the k-space data, are shown in Fig. 14. The back Fourier transforms of these RDFs, after setting lmGNiX*(r) equal to lmGNiX*(0) (Table 3) at r⩽rmin, are found to be in good agreement with the original scattering functions (see Fig. 13) from which they were obtained. The calculated real-space functions (Fig. 14) show three well pronounced peaks, which correspond to the first, third and fourth peaks, respectively, in the first-order difference functions, lmGNi(r) (see Fig. 8).
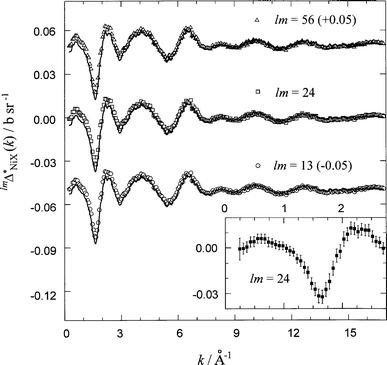 | ||
| Fig. 13 High-order difference functions, lmΔNiX*(k). Symbols are the data points and curves are the back Fourier transforms of the real-space lmGNiX*(r) functions after setting them to their low r-value limits, lmGNiX*(0) (full curves in Fig. 14). The inset shows the level of statistical noise (errors) in the experimental data at the high-order difference level. | ||
 | ||
| Fig. 14 High-order real-space difference functions, lmGNiX*(r) vs. r. The dotted curves are the Fourier transforms of the measured lmΔNiX*(k) functions of Fig. 13 and full curves are lmGNiX*(r) after the unphysical oscillations at r⩽1.85 Å have been set to their low r-value limits, lmGNiX*(0) (see Table 3). | ||
Next, the functions
 | (26) |
were fitted by the sum of three Gaussians, f1(r)+f2(r)+f3(r), each in the form of eqn. (18). The results of the fit are illustrated in Fig. 15, and the fitted parameters of the Gaussians are summarised in Table 7. Firstly, the Ni–Ni contribution is negligible due to low concentration of Ni2+ as compared to methanol atoms (see Tables 1 and 3). Secondly, since it is impossible for two nickel ions to be at such short distances where the first solvation shell of Ni2+ is observed, only the correlations of Ni2+ with O, C and D1–D3 atoms should be taken into account for further consideration.
![[lmGNiX*(r)−lmGNiX*(0)] functions fitted by a sum of three Gaussians represented by the functions, fi(r) of eqn. (18). The symbols correspond to the data, the dotted curves give the individual fitted Gaussians and the full curve gives their sum.](/image/article/2000/CP/a907829k/a907829k-f15.gif) | ||
| Fig. 15 [lmGNiX*(r)−lmGNiX*(0)] functions fitted by a sum of three Gaussians represented by the functions, fi(r) of eqn. (18). The symbols correspond to the data, the dotted curves give the individual fitted Gaussians and the full curve gives their sum. | ||
| Difference (lm) | |||
|---|---|---|---|
| Peak and its parameters | 1–3 | 2–4 | 5–6 |
| a rmax, σ and A are the mean position, half-width at half-maximum and area of a peak. NiO, NiC+NiD1 and NiD2,3 are the co-ordination numbers of the methanol atoms around the Ni2+ obtained by integration of the corresponding Gaussians over the ranges Δr1, Δr2 and Δr3, respectively. | |||
| 1st peak | |||
| rmax/Å | 2.057(1) | 2.056(1) | 2.057(1) |
| σ/Å | 0.103 | 0.103 | 0.103 |
| A/b | 0.0157 | 0.0150 | 0.0159 |
| NiO | 5.0(3) | 4.9(3) | 4.9(3) |
| Δr1/Å | 1.7–2.4 | 1.7–2.4 | 1.7–2.4 |
| 2nd peak | |||
| rmax/Å | 3.163(2) | 3.163(2) | 3.163(2) |
| σ/Å | 0.154 | 0.153 | 0.153 |
| A/b | 0.0154 | 0.0157 | 0.0157 |
| NiC+NiD1 | 10.2(6) | 10.1(6) | 10.1(6) |
| Δr2/Å | 2.6–3.7 | 2.6–3.7 | 2.6–3.7 |
| 3rd peak | |||
| rmax/Å | 4.000(7) | 3.999(8) | 3.999(7) |
| σ/Å | 0.213 | 0.218 | 0.218 |
| A/b | 0.0105 | 0.0109 | 0.0109 |
| NiD2,3 | 11(1) | 11(1) | 11(1) |
| Δr3/Å | 3.5–4.5 | 3.5–4.5 | 3.5–4.5 |
The first peak in lmG*NiX(r) functions is undoubtedly due to the Ni–O correlation. It is interesting to note that the position of this peak at 2.057 Å coincides for all the three RDFs within an accuracy of 0.001 Å (Table 7), thus resolving the discrepancy observed for this distance at the first-order difference level (see Table 4 and Section V.A). This distance is also in excellent agreement with the mean Ni–O distance of 2.05 Å in Ni(CF3SO3)2–methanol solution obtained from the recent EXAFS experiment.8
On the basis of the ab initio calculations performed (see Table 6 and Section V.C), one expects that the correlation of Ni2+ with hydrogens of the methyl group can be split into two types, Ni–D1 and Ni–D2,3, in accordance with Fig. 1. So, the second peak occurring at 3.163(2) Å (Table 7, Fig. 15) arises from a combination of Ni–C and Ni–D1 correlations, which can not be separated into individual contributions. The third peak at 4.000(8) Å, according to the spatial orientation of the methanol molecule with respect to the nickel cation (see Fig. 1), arises purely from the Ni–D2,3 correlations.
Following the above assignment of peaks and distances, the pair RDFs, gNiO(r), gNiC(r)+gNiD1(r) and gNiD2,3(r) calculated by using the fitted parameters from Table 7 are shown in Fig. 16. The pair RDFs, gNiHO(r) and gNiCl(r) obtained by using the second-order difference technique (see Section V.B) are also included in this figure. Fig. 16 clearly shows a high degree of overlap of the gNiCl(r) with both gNiO(r) and gNiHO(r). This is the reason why the positions of the first peaks (Table 4) in the first-order difference functions lmGNi(r) (Fig. 8) are shifted by 0.01–0.04 Å towards high r-values as compared to those obtained from high-order RDFs (Table 7). On the basis of similar arguments, the positions of the second peaks in the lmGNi(r) that reflect Ni–HO correlation are shifted towards the low r-values for CD3OD samples and, to the high r side for CD3OH samples, as compared to the rmax at 2.619 Å of gNiHO(r) (see Tables 4 and 5).
 | ||
| Fig. 16 The pair RDFs, gNiX(r) (full curves) describing the nearest-neighbour environment of Ni2+ in NiCl2–methanol solutions as obtained by using the second- and high-order difference techniques of neutron diffraction. The dotted curve is the gNiCl(r) pair RDF scaled as it contributes into the gNiO(r) and gNiHO(r) pair RDFs in the first-order difference functions, lmGNi(r) shown in Figs. 8 and 9. | ||
The co-ordination numbers obtained by integration of the fitted Gaussians, f1(r), f2(r) and f3(r), are listed in Table 7. Since the weighting factors lmWNiC and lmWNiD/3 are the same within 0.5% (Table 3), the integration of the f2(r) as an individual pair correlation function yields
 | (27) |
Similarly,
on the basis of the methanol molecule structure
and its spatial disposition in the Ni2+ solvation shell (Fig. 1), one can expect the value for the NiD2,3 co-ordination number to be twice that for the NiO. Our results of 4.9–5.0(3), 10.1–10.2(6), and
11(1) (Table 7) for the co-ordination numbers, NiO, NiC+NiD1
and NiD2,3, respectively, confirm the conclusion (see Section V.B) that the first co-ordination shell of Ni2+
is comprised of five methanol molecules and one chloride anion disposed in an octahedral arrangement around the central cation.
E. The orientation of the methanol molecule in the cation solvation shell
The sketch of Ni2+–methanol conformation optimised by ab initio calculations (Fig. 1) and the mean interatomic distances derived by using the second- and high-order difference techniques of ND, allow us to calculate the two angles, viz. ∠NiOC and ∠NiOHO,| cos(∠NiOC)=(rNiC2−rNiO2−rCO2)/(−2rNiOrCO), | (28) |
| cos(∠NiOHO)=(rNiHO2−rNiO2−rOHO2)/(−2rNiOrOHO). | (29) |
These angles, in fact, determine the orientation of the methanol molecule with respect to the co-ordinating ion. The distances rNiO=2.057 Å, rNiHO=2.619 Å and rNiC=3.163 Å were taken from Tables 5 and 7, whereas for rCO and rOHO, three different data sets were used: from (i) ab initio MP2/TZV** calculations, (ii) microwave gas-phase experiment30 and, (iii) ND liquid-phase experiments31 (Table 6). Calculated values of ∠NiOC and ∠NiOHO are summarised in Table 8 along with the values of ∠COHO derived by assuming that the Ni2+ lies within the symmetry plane of the methanol molecule. On the basis of a joint analysis of the structural parameters presented in Tables 6 and 8, two conclusions can be drawn.
| Angle/degrees | (a) | (b) | (c) |
|---|---|---|---|
| ∠NiOC | 122.4 | 129.7 | 130.38 |
| ∠NiOHO | 114.6 | 116.5 | 115.54 |
| ∠COHO | 123.0 | 113.8 | 114.08 |
First, the ∠NiOC of 122.4°, obtained by using the Ni–C and Ni–O distances from our NDIS experiments and intramolecular C–O distance from the MP2/TZV** ab initio calculations, is in good agreement with the one calculated theoretically, 125.9°. Worse agreement is, however, observed for the ∠NiOHO. This is, perhaps, because the ab initio calculations were performed only with one methanol molecule in the Ni2+–methanol complex. The presence of additional ligands around Ni2+ in the real solution will bring a change in the position of hydroxy hydrogen, and consequently, in the ∠NiOHO.
The second point to note is that all the three angles, i.e., ∠NiOC, ∠NiOHO and ∠COHO lie close to a value of 120°, which corresponds to sp2 hybridisation of the oxygen of methanol molecule with a plane-triangular geometry of the surrounding atoms, Ni, HO and C. In this case one can expect that residual lone-pair of the oxygen atom should occupy its 2px (2py) orbital situated perpendicular to the symmetry plane of the methanol molecule and the triangle formed by Ni, C and HO atoms. A strong preference for a triangular orientation of the methanol molecules surrounding Mg2+ in its octahedral solvation shell was also established by Tamura et al.32 from a molecular dynamics simulation of MgCl2 solution in methanol.
VI. Conclusions
A detailed investigation of the Ni2+ co-ordination shell at an atomic level in NiCl2–methanol solution has been carried out by combining neutron diffraction experimental results with ab initio calculations of Ni2+–methanol complex. The first-, second- and high-order difference methods with isotopic substitution on nickel, chlorine and hydroxy hydrogen were applied to obtain pair radial distribution functions of Ni2+. Special attention was paid to properly normalise the various scattering patterns, which is crucial to obtain accurate values of the structural parameters such as inter-atomic distances and co-ordination numbers. The main results of the present work can be summarised as follows.
For the first time in non-aqueous media, it has been shown that chloride ion penetrates the nickel(II) solvation shell, forming contact Ni–Cl complex (‘
contact ion pair’) with a mean separation
distance of 2.348(2) Å and co-ordination number of 0.72(5)
in NiCl2–methanol solution. We also established that because of this inner sphere complexation, it is not possible to derive
exact values of mean Ni–O distance and NiO
co-ordination number
purely from the first-order difference functions. By applying the second and high-order difference methods of ND we found, with high accuracy, the mean Ni–O distance of 2.057(1) Å and NiO value of 5.0(3). The integration of gNiHO(r) pair RDF with a maximum at 2.619(6) Å gave a co-ordination number of 5.4(5) for the NiHO.
On the basis of the ab initio analysis of the interatomic distances in Ni2+–methanol complex, the peaks occurring ca. 3.16 and 4.00 Å in the first- and high-order real-space difference functions have been assigned to Ni–C+Ni–D1 and Ni–D2,3 correlations, where D1,2,3 are the three methyl hydrogens split spatially into two different environments (D1 and D2,3) around Ni2+. The integrations of these peaks confirm that the Ni2+ is co-ordinated to five methanol molecules. By taking into account the electronic structure of the nickel(II) ion, it was concluded that the Ni2+ has a 6-coordinated octahedral structure of its nearest-neighbour environment.
The theoretically calculated and experimental interatomic distances have been used to derive ∠NiOC and ∠NiOHO. These angles are found to be consistent with sp2-hybridisation of the hydroxy oxygen and triangular-planar orientation of the methanol molecules surrounding Ni2+ in its solvation shell.
The above results have unequivocally demonstrated the power of using complementary techniques in resolving structural details of non-aqueous electrolyte solutions at a microscopic level. In a forthcoming paper, we shall present the results of a similar neutron diffraction study of Cl− co-ordination shell in NiCl2–methanol solutions.
Acknowledgements
We are grateful to Dr R. A. Howe for providing us with the isotopes, Dr H. E. Fischer (ILL) for help with the NDIS experiments, Dr V. V. Ivanov for helpful discussions regarding ab initio calculations and, the EPSRC, UK for support. One of us (O.N.K.) wishes to thank the Royal Society/NATO for the award of a post-doctoral fellowship and the University of Abertay Dundee for hosting his visit.References
- H.-J. Gores and J. Barthel, Pure Appl. Chem., 1995, 67, 919 CAS.
- G. W. Neilson and J. E. Enderby, J. Phys. Chem., 1996, 100, 1317 CrossRef CAS.
- G. W. Neilson and A. K. Adya, Annu. Rep.–R. Soc. Chem. Sect. C, 1997, 93, 101 Search PubMed.
- G. Sesê and J. A. Padro, J. Chem. Phys., 1998, 108, 6347 CrossRef CAS.
- D. H. Powell and G. W. Neilson, J. Phys. Condens. Matter, 1990, 2, 5867 CrossRef CAS.
- R. Ripan and I. Ceteanu, Chimia Metalelor, Editura Didactica Si Pedagogica, Bucharest, Romania, 1969, vol. II. Search PubMed.
- P. B. Lond, P. S. Salmon and D. C. Champeney, J. Am. Chem. Soc., 1991, 113, 6420 CrossRef CAS.
- Y. Inada, H. Hayashi, K. Sugimoto and S. Funahashi, J. Phys. Chem. A, 1999, 103, 1401 CrossRef CAS.
- D. G. Montague, I. P. Gibson and J. C. Dore, Mol. Phys., 1981, 44, 1355 CAS.
- G. L. Squires, Introduction to the Theory of Thermal Neutron Scattering, Cambridge University Press, Cambridge, 1978. Search PubMed.
- P. A. Egelstaff, An Introduction to the Liquid State, Oxford University Press, Oxford, 1992. Search PubMed.
- A. K. Soper, G. W. Neilson, J. E. Enderby and R. A. Howe, J. Phys. C: Solid State Phys., 1977, 10, 1793 Search PubMed.
- C. Cossy, A. C. Barnes, J. E. Enderby and A. E. Merbach, J. Chem. Phys., 1989, 90, 3254 CrossRef CAS.
- J. E. Enderby, D. M. North and P. A. Egelstaff, Philos. Mag., 1966, 14, 961 Search PubMed.
- G. W. Neilson and J. E. Enderby, Proc. R. Soc. London, Ser. A, 1983, 390, 353 Search PubMed.
- Y. S. Badyal and R. A. Howe, J. Phys. Condens. Matter, 1996, 8, 3733 CrossRef.
- Yu. V. Karyakin and I. U. Angelov, Pure Chemical Substances, ‘‘Khimiya’’ Publishers, Moscow, 1974, p. 292. Search PubMed.
- M. Bertagnolli, P. Chieux and M. D. Zeidler, Mol. Phys., 1976, 32, 759.
- V. F. Sears, Neutron News, 1992, 3, 26 Search PubMed.
- D. H. Powell and G. W. Neilson, J. Phys. Condens. Matter, 1990, 2, 3871 CrossRef CAS.
- H. H. Paalman and C. J. Pings, J. Appl. Phys., 1962, 33, 2635.
- I. A. Blech and B. L. Averbach, Phys. Rev., 1965, 137, 1113 Search PubMed.
- J. E. Enderby and G. W. Neilson, in Water, A Comprehensive Treatise, ed. F. Franks, Plenum Press, New York, 1979, vol. 6, ch. 1. Search PubMed.
- G. Placzek, Phys. Rev., 1952, 86, 377 CrossRef CAS.
- J. G. Powles, Adv. Phys., 1973, 22, 1 CAS.
- W. H. Press, S. A. Teukolsky, W. T. Veterling and B. P. Flannery, Numerical Recipes in Fortran: The art of Scientific Computing, Cambridge University Press, Cambridge, 1992. Search PubMed.
- R. A. Robinson and R. H. Stokes, Electrolyte Solutions, Butterworths Scientific Publishers, London, 1959. Search PubMed.
- G. W. Neilson, in Water and Aqueous Solutions, ed. G. W. Neilson and J. E. Enderby, Adam Hilger, Bristol, 1986, p. 169. Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- R. M. Lees and J. G. Baker, J. Chem. Phys., 1968, 48, 5299 CrossRef CAS.
- A. K. Adya, L. Bianchi and C. J. Wormald , J. Chem. Phys., in the press. Search PubMed.
- Y. Tamura, E. Spohr, K. Heinzinger, G. Pálinkás and I Báko, Ber. Bunsen-Ges. Phys. Chem., 1992, 96, 147 Search PubMed.
Footnotes |
| † Permanent address: Department of Inorganic Chemistry, Kharkov State University, Kharkov, Svobody Square 4, 61077, Ukraine. E-mail: Oleg.N.Kalugin@univer.kharkov.ua |
| ‡ 1 b (barn)=10−28 m2 |
| This journal is © the Owner Societies 2000 |