Spectroscopy and photophysics of tropolone in condensed media
Lian C. T. Shoute, Valerie J. MacKenzie, Kenneth J. Falk, Hemant K. Sinha, Aaron Warsylewicz and Ronald P. Steer*
Department of Chemistry, University of Saskatchewan, 110 Science Place, Saskatoon, SK, Canada S7N 5C9. E-mail: steer@usask.ca
First published on UnassignedUnassigned22nd December 1999
Abstract
A systematic study of the spectroscopy and photophysics of tropolone in condensed media has been undertaken by measuring its UV–visible absorption, emission and emission–excitation spectra in a number of solvents of varying structure, at temperatures between 77 K and 295 K, and by measuring its quantum yields of emission, ϕem, and time-resolved emission decays as a function of temperature, T, and excitation wavelength, λex, in each solvent. In weakly interacting solvents, such as 3-methylpentane and perfluoro-2-n-butyltetrahydrofuran, the only species yielding significant fluorescence is the S1, 1(π,π*) state of the intramolecularly hydrogen-bonded neutral molecule. In acidic aqueous solution at pH <3, the emitting state is also 1(π,π*) S1, but the intramolecular hydrogen bond has been disrupted by the solvent. In basic aqueous solution at pH >8 the emitting species is the corresponding excited anion. In some solvents, emission from photochemical products prevents reliable measurements from being made at room temperature. Contrary to previous reports, no emission from solvent-stabilised 1(n,π*) excited states is observed. In glass-forming media the fluorescence quantum yields and the lifetimes of the emitting species increase in a sigmoidal fashion as the temperaure decreases, and are a factor of more than 100 greater at 77 K than at room temperature. The fluorescence quantum yields also decrease with increasing excitation energy in the UV, an effect which is most pronounced in more weakly interacting solvents at temperatures near room temperature. A mechanism is proposed. Both the spectra and the excited state temporal decays indicate that emission from short-lived, vibrationally unrelaxed S1 constitutes the majority of the fluorescence at room temperature, a small fraction of the total emission at 77 K, and an increasing fraction of the total emission with decreasing excitation wavelength. The variations of ϕem and lifetime with temperature are attributed to the effects of temperature-dependent solvent relaxation dynamics on the non-radiative decay, based on qualitative correlations between the observed parameters describing the effects in different media and known characteristics of the media, including their glass transition temperatures and polarity.
Introduction
Photon-initiated intramolecular and intermolecular excited state proton transfer reactions are intimately involved in such diverse processes as photoprotection by additives in paints and other coatings,1 the mode of action of light-induced anti-viral agents,2,3 the gain mechanism of proton transfer lasers,4 and the active processes in some molecular information and energy storage devices.5,6 Recently, tropolone (TRN, Scheme 1) has been used as a model chromophore for the systematic study of the effects of microsolvation and cluster size on both intramolecular and intermolecular excited state proton transfer.7 Tropolone and its simple derivatives are useful models for such studies because (i) TRN is a relatively small molecule with a rich but readily interpretable vibronic spectrum that is observable in both absorption and fluorescence emission, (ii) the vibronic spectrum of the cold, isolated molecule exhibits accurately measurable proton tunneling doublets whose splitting is a direct measure of intramolecular proton transfer rate and (iii) the magnitude of these tunneling doublet splittings is mode and substituent sensitive and is affected by solvation in van der Waals complexes. Unlike other model sytems8,9 for studying proton transfer, the TRN chromophore offers the advantage that its two tautomers (I and II with transition state, TS, Scheme 1) have identical structures so that dual emission is not expected. | ||
| Scheme 1 | ||
The jet-cooled fluorescence excitation spectra of the bare TRN molecule and several of its van der Waals complexes with rare gases, water, methanol and small non-polar molecules have been reported.10–13 A recent review7 notes that, in considering the effects of microsolvation on TRN, solvent species may be usefully categorized as (i) those which are primarily dispersively bound, and (ii) those which are hydrogen-bonded. In the former case, a red solvatochromic shift is observed and the first and second addend species are preferentially located above and below the plane of the seven-membered ring, whereas in the latter case, a blue shift is observed and the first addend species is bound in plane, partially disrupting the intramolecular hydrogen bond between the hydroxy and keto groups.
We have recently reported the first direct measurements of the excited state dynamics of the bare TRN molecule,14 and interpreted the results with the aid of semi-empirical calculations of the energies of its electronic excited states. This investigation showed that the fluorescent S1 state in the isolated bare molecule is of π,π* character, that its major decay path is non-radiative (the fluorescence quantum yield from the zero point level is ca. 0.06), that the radiationless relaxation rate increases rapidly with increasing vibrational energy in S1, and that there is no measurable dependence of the radiationless decay rate on the parity of each tunneling doublet. The available evidence indicates that, in the isolated molecule, intersystem crossing dominates S1's decay processes at v = 0 whereas internal conversion is responsible for the rapid increase in decay rate with increasing vibrational energy, in accord with Lim's ‘‘proximity effect’’ model.15
Curiously, more is now known about the spectroscopy and photophysics of tropolone and its derivatives in supersonic expansions than in bulk media. Numerical values of the excited state lifetimes and fluorescence quantum yields have been reported for tropolone in a variety of condensed media16–19 but are apparently inconsistent and do not present a coherent picture of the molecule's decay dynamics. The only definitive conclusions that can be drawn from the existing photophysical data for TRN in condensed media are that (i) like the gas phase, the quantum yields of S1–S0 fluorescence are small and radiationless relaxation dominates the decay of S1, and (ii) the triplet yield is large but the triplet states are short-lived and non-phosphorescent.
This paper presents the results of the first systematic study of the photophysics of TRN in condensed media. The data permit some parallels to be drawn between the behaviour of excited tropolone in condensed media and in supersonically expanded clusters.
Experimental section
Materials
Because tropolone is only weakly fluorescent in condensed media at room temperature, great care was taken to rigorously purify the solute and solvents in order to obtain meaningful results. Tropolone (Alpha) was either recystallized four times from dry c-hexane or recrystallized and sublimed, and was routinely used in freshly prepared solutions in order to minimize problems of photodecomposition during spectroscopic experiments.Many different solvents were employed initially in experiments at room temperature, but only those capable of forming low temperature glasses were used extensively thereafter. 3-Methylpentane (3MP, Aldrich) was passed through AgNO3-impregnated silica gel to remove fluorescent impurities and was dried by distillation from sodium metal. 2-Methyltetrahydrofuran (MTHF, Aldrich) was distilled from sodium metal under nitrogen; only freshly-distilled solvent was used to prepare solutions in order to minimize oxidation. Ethanol (EtOH, abs, Aldrich) was puified by distillation. Perfluoro-2-n-butyltetrahydrofuran (FTHF, TCI) was passed through freshly dried silica gel, and was degassed by freeze–pump–thaw cycles and sealed off under vacuum when clear glasses at 77 K were required.
The temperature dependence of the fluorescence of tropolone as a function of pH in aqueous solution was studied using 6.5 M aqueous LiCl as a solvent. Such solutions form a clear glass at 77 K, and are strongly buffered at high pH.20 Water obtained from a Millipore purification system was used in these experiments, and the pH was adjusted to between 0 and 13 using aqueous HCl and NaOH, respectively.
Instrumentation
Absorption measurements were taken with either a Cary 118 or a Cary 1E UV–visible spectrophotometer. Fluorescence emission spectra, emission excitation spectra and quantum yields were measured using a Spex Fluorolog 212 spectrofluorometer. Measurements with these instruments at temperatures below room temperature were obtained using a variable temperature Oxford Instruments cryostat equipped with a digital temperature controller. For temperatures at which the solutions were liquid, the sample was prepared in a 1 × 1 × 4 cm Suprasil quartz cuvette fused to an inlet tube which was compression-sealed into the cryostat and equipped with an inlet for purging air from the system using dry high purity argon. At temperatures at which the samples formed glasses down to 77 K, the liquid solution was sealed in a 6 mm id Suprasil quartz tube. Dry nitrogen gas was used as a heat exchange medium in this cryostat, and care was taken to ensure that the equipment had reached thermal equilibrium at each new temperature. A fresh sample was used at each temperature to avoid artefacts resulting from photochemical degradation of the sample in the spectrofluorometer.Fluorescence lifetimes were measured with a Spectra-Physics mode-locked, synchronously-pumped, cavity-dumped Nd: YAG dye laser combination using methods which have been described in detail previously.21 The mode-locked fundamental of the Nd:YAG laser (ca. 10.5 W cw) was frequency-doubled to give ca. 1.4 W at 532 nm for synchronously pumping a tunable R6G dye laser. The dye laser was cavity-dumped (Spectra-Physics Model 3290) at 4 MHz to give a train of ps pulses with an average power of ca. 90 mW in the fundamental at 620 nm. Frequency-doubling the cavity dumped output in α-BBO (Casix, Inc.) produced excitation wavelengths in the 310 nm range, coincident with the S4–S0 band system of tropolone. Fluorescence excitation and decay profiles were obtained by time-correlated single photon counting and were analyzed by methods described previously.21 Emission was observed through a polarizer set to the magic angle with respect to the plane of polarization of the excitation beam in order to avoid artefacts due to rotational diffusion.
Efforts were also made to measure the lifetimes of TRN excited directly in the origin of the S1–S0 absorption system by pumping with an argon-ion pumped dye laser, but they were unsuccessful. The excitation wavelength required to excite into the S1 origin is difficult to achieve by frequency doubling the output of a dye laser. Dyes which lase in the 740 nm range (required before frequency doubling) generally absorb rather weakly at the pump wavelength of 514.5 nm. With our equipment we were only able to achieve laser action in these dyes when pumping with a 2 W cw beam. No lasing was achieved with a mode-locked 1 W pump source, and thus lifetime measurements could not be made at the desired 370 nm excitation wavelength.
Methods
Quantum yields of fluorescence were measured by the well-established relative method using quinine sulfate in 0.5 M aqueous H2SO4 (ϕf = 0.54622) as a standard and correcting for any differences between the refractive indices, n, of the standard solution and the sample solution using the appropriate ‘‘n2 ’’ factor.23 Solutions were prepared so that the absorbances of the sample and standard solutions at room temperature were nearly the same at the wavelength of excitation and were less than 0.1 to avoid geometric artefacts. The intensities of emission of the sample in the liquid and clear glassy states were measured at temperatures down to 77 K in rectangular quartz cuvettes. The measured sample emission intensities were converted to quantum yields by comparing them with the emission intensity of the quinine sulfate standard at room temperature under otherwise identical conditions. A correction to the sample emission intensity accounting for the change in density, d, and refractive index with temperature must be made in order to obtain exact emission quantum yields at T<298 K. This correction (the ratio of d/n2 for the sample at T to d/n2 for the standard at 298 K) changes the uncorrected quantum yield by less than 10% at 77 K.Excitation of tropolone throughout the UV region results in photochemical decomposition. The rate of this decomposition could be followed by measuring the absorbance of the starting solution as a function of time, and was found to be particularly rapid at room temperature in some organic solvents. Although no attempt was made to identify the final products, it was observed that some of them were fluorescent, and this prevented the measurement of accurate quantum yields of tropolone fluorescence in some solvents at room temperature. To minimize this problem, fresh solution was used for every critical measurement.
All spectra have been corrected to account for the variation of the sensitivity of the measuring instruments with excitation or emission wavelength.
Results and discussion
In order to provide a database for the systematic analysis of the spectroscopy and photophysics of tropolone in condensed media, UV–visible absorption, emission and emission–excitation spectra were measured in a number of solvents of varying structure and polarity, and quantum yields of emission and time-resolved emission decays were measured as a function of temperature and excitation wavelength in several selected solvents.At fixed excitation wavelength in a given solvent the emission quantum yields, ϕem, generally increase in a sigmoidal fashion with decreasing temperature, as shown for example in Fig. 1 for TRN in FTHF. Values of ϕem at the two extremes of temperature, 77 K and 295 K, and the approximate temperature at the inflection point of the sigmoidal curve, T1/2 , are given in Table 1 for each of the solvent systems investigated. The quantum yields also increase with increasing excitation wavelength in fluid media, and reach a maximum when excited at wavelengths near the origin of the S1–S0 transition (vide infra).
 | ||
| Fig. 1 Quantum yield of fluorescence of TRN in FTHF excited at 352 nm as a function of temperature. | ||
| Solvent | λex/nm | ϕem(77 K) | ϕem(295 K) | T1/2/K | TGa/K | Eab/cm−1 |
|---|---|---|---|---|---|---|
| a Glass transition temperatures from refs. 24 and 25.b Apparent activation energies for the competing radiative (r) and radiationless (nr) processes leading to emission i.e., Ea = ΣEa,nr − Ea,r.c Impurity fluorescence dominates at room temperature. | ||||||
| 3MP | 360 | 0.06 | 2.3 × 10−4 | ca. 100 | 77 | ca. 700 |
| MTHF | 360 | 0.06 | 2.4 × 10−4 | 110 | 90 | 600 |
| Ethanolc | 360 | 0.23 | — | 114 | — | 520 |
| FTHF | 340 | 0.06 | 1.2 × 10−4 | 140 | — | 1030 |
| 6.5 M LiCl, pH = 0 | 340 | 0.14 | 3.8 × 10−4 | 188 | 140 | 1230 |
| 6.5 M LiCl, pH = 9 | 360 | 0.21 | 1.6 × 10−3 | 183 | 140 | 1500 |
| H2O, pH = 0 | 330 | — | 1.2 × 10−4 | — | — | — |
| H2O, pH = 9 | 340 | — | 1.4 × 10−4 | — | — | — |
Many of the spectra and most of the quantum yields and fluorescence decay times reported here are significantly different from most of those in the existing literature.16–19 For example, only the value of ϕem = 0.18 obtained by Croteau and Leblanc16 for TRN in EPA glass at 77 K agrees reasonabl y well with the data in Table 1. Even in this case, however, our attempts to reproduce the previous result were complicated by the observation of an emission intensity which depended on the time of exposure of the sample to excitation light in the spectrofluorometer, likely indicative of the photochemical production of interfering fluorescent substances. A detailed analysis of such discrepancies will not be attempted, except to note that previous authors have probably not appreciated the functional dependence of TRN's decay characteristics on solvent structure, temperature and excitation wavelength, and the need for rigorous solute and solvent purity.
(i) Properties of TRN in weakly interacting solvents
The UV–visible absorption, emission and emission excitation spectra of TRN in 3-methylpentane (3MP) at room temperature are shown in Fig. 2, and in a 3MP glass at 77 K in Fig. 3. These spectra are similar to their counterparts in other weakly interacting solvents, perfluoro-n-hexane (PFH), and perfluoro-2-n-butyltetrahydrofuran (FTHF) (not shown). Two broad, moderately strong features (εmax = 5000–6000 dm3 mol−1 cm−1) with maxima near 355 and 320 nm are observed in the absorption spectra of TRN in these solvents. Some vibrational structure, including a relatively strong feature near 370 nm, can be observed and acts as a partial fingerprint of the absorbing states. The absorption spectra are similar to those for TRN in cyclohexane reported by Bréhéret and Martin18 but the emission excitation spectra are completely different. The long-wavelength bands observed by Bréhéret and Martin in their excitation spectra and assigned to emission from a low-lying 1(n,π*) state are not observed when pure aliphatic hydrocarbons or other weakly interacting solvents are employed and the extent of TRN photochemical decomposition is minimized.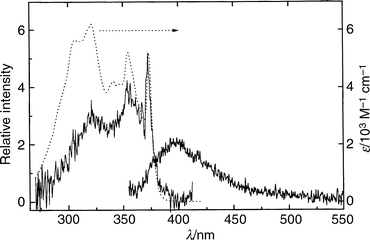 | ||
| Fig. 2 Absorption (···), emission (———, right; λex = 320 nm) and emission excitation (———, left; λem = 416 nm) spectra of TRN in 3MP at room temperature. The intensity of the excitation spectrum has been normalized to the 370 nm band of the absorption spectrum. | ||
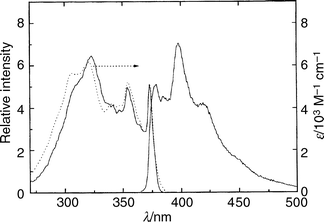 | ||
| Fig. 3 Absorption (···), emission (———, right; λex = 320 nm) and emission excitation (———, left; λem = 416 nm) spectra of TRN in 3MP glass at 77 K. The intensities of the emission and emission excitation spectra have been normalized to the 370 nm band of the absorption spectrum. | ||
In order to determine the extent to which TRN might be self-associated in these weakly interacting solvents, the absorption and emission spectra were measured as a function of the concentration of TRN up to ca. 3 × 10−3 M at room temperature. Apart from the expected changes in intensity, the spectra were independent of TRN concentration; there is no evidence of solute self-association at the concentrations employed. This observation is in agreement with the results of light scattering experiments on TRN in several non-polar solvents,26 but in disagreement with a suggestion based upon polarization measurements and MO calculations.27 Despite the lack of evidence of self-association, most spectra, quantum yields and time-resolved emission decays were measured in very dilute (⩽10−5 M) solutions to avoid the possibility of forming significant amounts of dimer.
Semi-empirical INDO-DSCI calculations14,28 indicate that two 1(π,π*) states (S1 and S4) and two 1(n,π*) states (S2 and S3) are accessible at these energies in the isolated molecule. The calculated oscillator strengths for the S1–S0 and S4–S0, π*←π, transitions are 0.107 and 0.089, respectively, while the S2–S0 and S3–S0, π*←n, transitions have much smaller values (0.0003 and 0.002, respectively) because they are electric dipole forbidden. Based upon the experimental absorption spectra, the magnitudes of the molar extinction coefficients and the results of the semi-empirical calculations, the two broad band systems in the near UV are assigned to the S1–S0 and S4–S0 π*←π transitions. The two π*←n transitions will be much weaker and, according to the calculations, should lie at energies between those of S1–S0 and S4–S0. The S2–S0 and S3–S0 transitions are therefore expected to be buried beneath the much more intense π*←π bands in the absorption spectra of the isolated molecule.
The fluorescence emission spectra of TRN in the three most weakly interacting solvents employed (3MP, FTHF and PFH) are almost identical; the same is true of the emission excitation spectra of TRN in the same media (cf. Figs. 2 and 3). The wavenumbers and relative intensities of the bands in the normalized emission spectra are almost independent of temperature at the low resolution employed. The spectra do broaden with increasing temperature, however, and lose much of their vibrational structure at room temperature. Allowing for differences in resolution of the spectrophotometers, the fluorescence spectra at low temperature are a reasonable mirror image of the absorption spectra in each case and the wavenumbers of the bands in each excitation spectrum coincide with those in the corresponding absorption spectrum, indicating that the state produced on absorption is responsible for a preponderance of the emission. The strong feature near 370 nm, which coincides in the absorption, excitation and emission spectra of TRN in these weakly interacting solvents, is assigned to the envelope of the electronic origin of the S1–S0 transition and its sequence bands, in agreement with the origin observed in the LIFE spectrum of the jet-cooled species7 (27018 cm−1; 370.1 nm). The cold isolated molecule is known to be intramolecularly hydrogen-bonded and to undergo fast proton tunneling between two identical tautomeric structures. Therefore the spectra in these weakly interacting solvents suggest that the absorption is due to the intramolecularly hydrogen-bonded ground state of the neutral molecule, and that the fluorescent species is the corresponding S1 state of 1(π,π*) character. There is no evidence of emission from the 1(n,π*) state as reported by Bréhéret and Martin.18
Comparisons of the absorption and normalized excitation spectra of TRN in these solvents at room temperature show that the relative intensity of the excitation spectrum decreases with decreasing wavelength compared with the absorption spectrum (cf. Fig. 2) in each case, but extends further to the blue compared with that of a room temperature vapour phase sample (not shown) or of cold isolated TRN in a supersonic expansion.7 These comparisons, together with the fact that the quantum yields of emission at all temperatures and excitation wavelengths are much less than 1 (cf. Table 1), indicate that (i) S1, of 1(π,π*) character, is the only state responsible for significant emission irrespective of the state initially populated, (ii) fast internal conversion, Sn (n = 2,3,4)→S1 in one or several sequential steps, is responsible for populating S1 from higher states, (iii) rapid radiationless processes also dominate the decay of S1 at all temperatures, but with increasing rates at higher temperatures, (iv) the rates of all radiationless processes which compete with S1→S0 fluorescence increase with increasing excitation energy, (v) vibrational relaxation in these weakly interacting solvents is not sufficiently rapid at room temperature to thermalize the fluorescent state even at modest initial vibrational energies, and (vi) ϕem is a much weaker function of excitation wavelength at 77 K than at room temperature.
(ii) Properties of TRN in water
In order to measure the properties of the ground and excited states of the neutral TRN molecule and its corresponding anions in a polar protic solvent medium, TRN was dissolved in water and either acidified with HCl or made basic with NaOH. The values of the ground and excited state pKa of TRN have been reported18 to be 6.7 and 2.2, respectively. Thus TRN should be present as the undissociated neutral molecule in both the ground and excited states under the acidic conditions employed (pH≈0) and as the anion in both the ground and excited states in the aqueous base at pH≈9. For measurements over the whole range of temperatures from 77 K to 298 K, 6.5 M LiCl was used to provide an aqueous solvent medium which would form a glass. Large concentrations of dissolved salts such as LiCl have the effect of disrupting the normal structure of water, and are observed here to increase the quantum yield of TRN fluorescence relative to water itself at T>273 K. Representative spectra at 77 K are shown in Fig. 4. The spectra taken at room temperature substantially reproduce those previously reported for TRN in acidic and basic aqueous solutions.18 | ||
| Fig. 4 Emission (right) and normalized emission excitation (left) spectra of TRN at pH 9 in 6.5 M aqueous LiCl glass at 77 K (———) and at pH 0 in 6.5 M LiCl glass at 77 K (···). The intensities of the emission and emission excitation spectra have been normalized to the same intensity in the origin. | ||
At room temperature the fluorescence quantum yield is small in both acid and base (<10−3, Table 1) and the spectra are poorly resolved, whereas at 77 K the quantum yields approach 0.14 for the excited neutral and 0.21 for the excited anion, and the spectra are quite revealing. Note in Fig. 4 that the undissociated acid at 77 K has an emission excitation spectrum which is similar to those of TRN in other condensed media at 77 K, except that the spectrum is shifted by ca. 20 nm to the blue so that the origin lies near 350 nm. This blue shift is consistent with the behaviour of undissociated TRN in water clusters of increasing size,7,12,13 and is a result of the disruption of the intramolecular hydrogen bond by strongly hy drogen-bonding solvents. The small red shift normally associated with π*←π transitions of aromatic molecules in more polar solvents is not seen here because it is swamped by the much larger effects on tropolone's intramolecular hydrogen bonding.
The vibrational structure of the emission spectrum of TRN in strongly acidic media is poorly resolved, as expected of a strongly solvated species, but the envelope of the emission itself is also blue-shifted by ca. 20 nm compared with that observed in poorly solvating media (cf. Figs. 2 and 3). The species responsible for the spectra taken at low pH are clearly assignable to the undissociated molecule, both in absorption and emission, but the position of the spectrum suggests that the intramolecular hydrogen bond of TRN has been completely disrupted and imparts little or no stability to the neutral species in this polar protic medium.
The emission and emission excitation (absorption) spectra of TRN in base (Fig. 4) are displaced ca. 30 nm to the red compared with those of the undissociated molecule observed in aqueous acid. Such displacements are common features of the spectra of acid–base pairs of aromatic alcohols in polar condensed media, and these spectra are completely consistent with a simple Förster cycle29 for TRN given its values of pKa and pKa*. Note that the emission excitation and absorption spectra consist of two broad features in the 300–400 nm region, assignable to two electric dipole allowed π*←π transitions in the anion, which are the counterparts of the S1–S0 and S4–S0 transitions in the undissociated molecule. Note also that the S1–S0 transition of the anion has a value of εmax which is substantially larger than that of the neutral (ca. 10 × 103 M−1 cm−1vs. ca. 6 × 103 M−1 cm−1). It is possible that the larger oscillator strength of the S1–S0 transition in the anion is at least partly responsible for its higher quantum yield of emission compared to the neutral.
(iii) Effect of excitation wavelength
The fact that the normalized emission excitation spectra of undissociated TRN at room temperature decrease in intensity more rapidly with decreasing wavelength than the corresponding absorption spectra in all solvents suggests that the rate of radiationless relaxation of the excited states increases with increasing excitation energy. In order to investigate this phenomenon more thoroughly, the quantum yields of emission as a function of excitation wavelength, ϕem(λex), were measured in the near UV region. The results for TRN in 3MP at room temperature are shown in Fig. 5. Qualitatively similar results were found for TRN in all other solvents employed, including acidic and basic aqueous media. | ||
| Fig. 5 Fluorescence quantum yield from TRN in 3MP at 298 K as a function of excitation wavelength. | ||
Note that at room temperature the quantum yields are small at all excitation wavelengths and the results are somewhat scattered because of this. Nevertheless, a clear trend of decreasing fluorescence quantum yield with increasing excita tion energy may be observed; the quantum yield is a maximum at excitation wavelengths near the electronic origin of the S1–S0 system at 370 nm, and within experimental error decreases monotonically with decreasing λex over the Sn (n = 2,3,4)–S0 absorption bands. Thus the quantum yields quantitatively reflect the differences in the relative intensities of the absorption, Iabs(λex), and corrected emission excitation spectra, Iex(λex), as a function of excitation energy, in accordance with the well-known relationship Iex(λex) = Iabs(λex)·ϕem(λex). The same effect is observed at 77 K in these weakly-interacting solvents, but the quantum yields of emission are much larger and the variation in ϕem as a function of λex is smaller. Again the trend in ϕem(λex) quantitatively reflects the differences in the ratio Iex(λex)/Iabs (λex); the decrease in ϕem(λex) with decreasing λex is considerably smaller at 77 K than at room temperature.
(iv) Mechanism
The temperature and excitation wavelength effects on the quantum yields of fluorescence of undissociated, intramolecularly hydrogen-bonded TRN, such as those found in weakly interacting solvents at all temperatures, may be treated quantitatively by considering the following simple mechanism. | (1) |
| Sn′(n = 2,3,4)→S1′kicSn (2) | (2) |
| S1′→S1thkth (3) | (3) |
| S1→S0 + hνfkr | (4) |
| S1→S0 + heat kicS1 | (5) |
 | (6) |
 | (7) |
Intermolecular processes, such as reaction with oxygen or concentration quenching, are not considered since neither oxygen nor TRN concentration in the range employed (<10−3 M) has any effect on the quantum yields. Although the nature of the photoproducts was not investigated in the present study, the rate of net photochemical consumption of the starting material could be followed by monitoring the changes in the absorbance of the TRN solutions. These rates, kpd, are faster in non-polar than in polar solvents and decrease with decreasing temperature in a given solvent.
In this mechanism, it is understood that S1 represents both unrelaxed S1 (S1′) or thermalized S1 (S1th) and that the rates of processes (2)–(7) are all functions of the vibrational energy content of the excited singlet states involved and of the extent of relaxation of the solvent in the vicinity of the initially excited solute. Generally these rates will increase with increasing vibrational energy content of the excited solute and will also change with the extent of solvent relaxation during the lifetime of the excited solute, although not all in the same functional fashion.
Intersystem crossing (isc) may occur from upper singlet states, but for simplicity is considered here only to involve S1 (step 6). The lowest triplet state of TRN lies near 201 kJ mol−1 and is reported to be non-phosphorescent even in glassy media at 77 K.16 We also have found no long-lived or oxygen-sensitive emission in any solvent at any temperature, and therefore confirm these results. We assume that intersystem crossing to the triplet manifold may be a major relaxation pathway for S1, particularly since first-order spin–orbit coupling between S1 and nearby 3(n,π*) states would be expected to result in a large value of kiscS1. However, it is reasonable to assume that the intersystem crossing process(es) will be irreversible and that triplet chemistry does not affect the relaxation of molecules in the excited singlet manifold.
(v) Effect of temperature
The effect of temperature on the fluorescence quantum yields at fixed excitation wavelength are qualitatively similar for both undissociated TRN and TRN anion in all solvents, as exemplified by the cases of the neutral in FTHF, Fig. 1, and the anion in 6.5 M LiCl at pH 9, Fig. 6. Note the sigmoidal shapes of these curves, starting from a low plateau at high temperatures and rising to a higher plateau at low temperatures. (The plateau in FTHF is expected to become distinct only at T<77 K.) The data which characterize these curves are given in Table 1. Similar sigmoidal shapes are observed for the anion and for the neutral in weakly interacting solvents, except that the values of T1/2 are different in different media. The sigmoidal shape of ϕem(T) therefore cannot be attributed to changes in the degree of dissociation of the TRN as a function of temperature.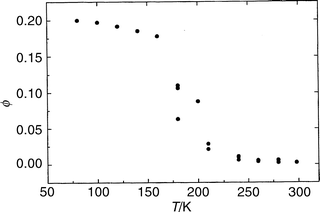 | ||
| Fig. 6 Quantum yield of fluorescence of the TRN anion in 6.5 M aqueous LiCl at pH 9 as a function of temperature. | ||
The above mechanism yields the following relationship between the quantum yields and the decay parameters for S1 at a fixed excitation wavelength;
| Σknr/kr = 1/ϕem − 1 |
In order to place the effect of temperature on a more quantitative basis, log10(1/ϕem − 1) was plotted vs. T−1 for each solvent system. The slopes of linear portions of each plot can be used to obtain the sums and differences of the apparent activation energies for the competing radiative and radiationless processes leading to emission, i.e., ΣEa,nr − Ea,r, in each distinct temperature regime. A typical plot is shown in Fig. 7 for TRN anion in 6.5 M LiCl. Three temperature regimes are observed over the entire temperature range. The quantity (ΣEa,nr − Ea,r) is less than 100 cm−1 both at low temperature (near 77 K) and at high temperature (T>150 K in basic 6.5 M LiCl), and in the intervening temperature range takes on the values listed in Table 1.
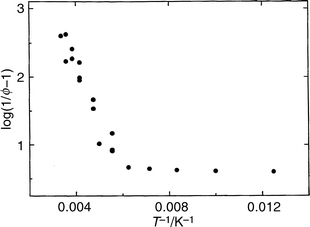 | ||
| Fig. 7 Plot of log10(1/ϕem − 1) vs. T−1 (see text) for TRN anion in 6.5 M aqueous LiCl at pH 9. | ||
Given that a large fraction of excited TRN decays by radiationless processes at all temperatures in all solvents, and that radiative rate constants are relatively insensitive to large changes in temperature, it is reasonable to expect that the observed variations in Σknr/kr are due primarily to changes in the overall rate of radiationless decay of S1 with temperature, i.e., to changes in Σknr. The low temperatures over which the large changes in emission yield occur suggest that dynamic solvent effects are responsible.30 Such dynamic solvent effects have been quantitatively investigated for the phosphorescent triplet states of several aromatic probe molecules in glassy media.31 It was not our intent to examine solvent dynamics at the beginning of this study, but rather to rationalise the disparate results of investigations of TRN photophysics obtained by us and others. Nevertheless, the present results appear to offer an opportunity to examine solvent dynamics in a hydrogen-bonded system on a time scale which is much faster than the millisecond to second time scale which applies to long-lived triplets. We therefore intend to measure the time-resolved emission spectra and decay kinetics of TRN as a function of temperature in these systems in future experiments and to quantify the solvation processes involved. For now, the following qualitative desciption of the mechanism appears to fit the existing data.
First, the temperatures at which the large changes in Σknr occur in various media, as given by the observed values of T1/2 (Table 1), increase with increasing glass transition temperature, TG (Table 1), and with increasing values of solvent polarity parameters such as the normalized Dimroth parameter ETN.31 Qualitatively, the same effect is observed on the variation of the measured triplet energies and decay times of highly phosphorescent aromatic compounds in various glassy media,24 except that the temperatures of ‘‘solvation ’’ (the equivalent of T1/2 measured here) are only slightly higher than TG (ca. 4 K higher in the case of quinoxaline triplets in various organic glasses24). Here the values of T1/2 are up to ca. 45 K higher than TG, suggesting that the shorter lifetime of the S1 state of TRN requires the more fluid medium that a higher temperature would provide to effect faster relaxation of the solvent, i.e., the viscosity of the medium must be lower if solvent relaxation is to have an effect on the dynamics of the shorter-lived excited species. The mechanism for increasing the radiationless transition rate by faster solvent relaxation as the temperature increases beyond TG is unclear, but could involve (i) tuning a 3(n,π*) state into resonance with the fluorescent S1 state of 1(π,π*) configuration to permit fast intersystem crossing by a first-order spin–orbit coupling mechanism,32 (ii) tuning a 1(n,π*) state into near resonance with S1 to permit fast internal conversion (ic) by the ‘‘proximity effect’’ mechanism described by Lim,15 or (iii) relaxation processes which disrupt the intramolecular hydrogen bond. Since the same effect is seen in both non-polar and polar solvents and in both the anion and the neutral in aqueous solution, the last of these mechanisms seems unlikely. The fact that the apparent activation energies (Table 1) for the solvent reorganization process at tempe ratures near T1/2 are much smaller than those observed at lower temperatures for longer-lived triplet states24 may suggest that only the translational component of the solvent relaxation is being revealed in these measurements.
(vi) Excited state temporal decays
When the electronically excited solute–solvent system is at thermal equilibrium, the lifetime of the emitting S1 state is given by the inverse of the sum of the rate constants for steps (4)–(7) inclusive;| τ = (kr + kicS1 + kiscS1 + kpdS1)−1, | (8) |
whereas the sum of the first-order rate constants for steps (5), (6), and (7) constitutes the overall rate constant for non-radiative decay;
| Σknr = kicS1 + kiscS1 + kpdS1. | (9) |
Since each of the rate constants in eqn. (8) and (9) is some function of the vibrational energy content of the excited species, the disposition of this energy among the available vibrational states, and the extent of relaxation of the solvent, both the emission quantum yields and rates of temporal decay of the fluorescent species are solvent and temperature dependent. Since all higher states will decay to vibrationally excited S1′, directly or indirectly, by the processes Sn(n = 2,3,4)→S1′, the vibrational energy content of S1′ will increase with increasing n leading to an increased rate of radiationless decay of the system to the ground state with decreasing excitation wavelength. The measured temporal decay profile of the ensemble of fluorescing species will be expected to be mono-exponential only when the ensemble is at thermal equilibrium with its surroundings.
The fluorescence decay times of TRN could only be measured with our equipment using excitation wavelengths which fall in the S4←S0 absorption system near 310 nm. As a result, contributions to the emission due to vibrationally unrelaxed TRN were observed, and the resulting temporal emission profiles had to be fit using bi-exponential or multi-exponential decay functions. At low temperatures, the decays could be modelled successfully using a bi-exponential decay function of the form
 | (10) |
where the relationship between the parameters in eqn. (10) and the fraction of the emission attributable to each species is given by
| Fi = aiτi/Σajτj | (11) |
A typical time-resolved emission profile of TRN in 3MP at 81 K together with the computed fit to the above bi-exponential function is shown in Fig. 8. The computed fitting parameters for TRN's temporal decay in 3MP as a function of temper ature are given in Table 2. Note that at temperatures less than ca. 100 K a bi-exponential decay function provides a reasonable, if not altogether satisfactory description of the decay. The longer-lived component has an average lifetime of 2.06±0.10 ns and contributes 91±2% of the total emission whereas the minor component has an apparent average lifetime of ca. 385±20 ps. At fixed low temperature these parameters are independent of emission observation wavelength over the entire emission spectrum. (The short-lived minor component of the emission at T<100 K is almost certainly not properly represented by one, single-exponential function; the bi-exponential function only gives a satisfactory fit because the second component is quite short-lived and constitutes only a small fraction of the total emission.) As the temperature increases, a bi-exponential decay function no longer provides a satisfactory description of the emission decay (cf. values of χ2, Table 2). Nevertheless, qualitatively the lifetime and fraction of the long-lived component(s) decrease, and the short-lived components increase in proportion until they dominate at room temperature.
 | ||
| Fig. 8 Log10 of the fluorescence intensity (number of counts) vs. time for TRN in 3MP at 81 K, excited at 310 nm. The number of counts in the peak channel is approximately 104. The lower line is the instrument response function and the upper lines are the best fit between the data and a bi-exponential decay curve with the parameters listed in Table 2. The bottom panel is the distribution of weighted residuals. A fit of the same data to a monoexponential decay function resulted in a significantly larger χ2. | ||
The longer-lived species has a lifetime which exhibits the same sigmoidal behaviour as a function of temperature as ϕem(T), and the ratio ϕem(T)/τ2(T)=kr remains approximately constant at ca. 2 × 107 s−1 over the entire temperature range. The latter value is consistent with the value of the radiative rate constant of the S1 state of tropolone derived from the oscillator strength of the S1–S0 transition in absorption. It is therefore reasonable to assign the longer-lived major component to vibrationally thermalized TRN in its S1 state (produced either directly or indirectly from S4 by internal conversion and vibrational relaxation). The large variations in the quantum yield and lifetime with temperature in glassy media can therefore be reasonably attributed to changes in the rate of non-radiative decay of the excited state associated with relaxation of the surrounding solvent. In the 3MP glass at low temperature, the lifetime of this vibrationally thermalized but solvent unrelaxed ensemble is τ = 2.06 ns and the fluorescence quantum yield is 0.04 (for TRN in 3MP at 77 K when excited at 310 nm). For comparison, the mono-exponential lifetime of isolated TRN excited to either of its lowest tunneling doublets in a supersonic expansion is 1.2 ns.14
At higher temperatures the average thermal energy of the vibrationally equilibrated ensemble increases, the solvent relaxes at much faster rates, and the lifetime of the ensemble of excited TRN species decreases. We speculate that the shorter-lived component(s) observed in the time-resolved decays can be assigned to the ensemble of vibrationally hot TRN molecules. Because a major component of the emission at higher temperatures comes from vibrationally unequilibrated species, a bi-exponential emission decay function no longer provides a satisfactory description of the temporal emission profile. The emission spectra of TRN in 3MP at room temperature provide confirmation of the presence of vibrationally hot emission. Note, in Fig. 2, the loss of vibrational structure and the onset of emission at shorter wavelengths than the 370 nm origin.
(vii) Properties of TRN in other media
The spectra of TRN in methyltetrahydrofuran at 77 K and room temperature are collected in Figs. 9 and 10, respectively. The spectra of TRN in ethanol (not shown) are similar except that the absorption spectra at all temperatures exhibit a distinct additional weak feature with an onset near 400 nm. The spectra of TRN in MTHF at 77 K closely resemble those of TRN in 3MP, and are clearly assignable to absorption and emission from neutral TRN which is intramolecularly hydrogen-bonded. (Note, however, the very weak absorption to the red of the TRN origin.) The absorption spectrum of TRN in MTHF at room temperature is similar to that at 77 K, but the emission and emission excitation spectra at room temperature are very different from those at 77 K. Note particularly that the FWHM of the emission spectrum of TRN in MTHF at room temperature is much larger than those of the other emission spectra, suggesting that more than one emitting species may be present. These effects are more pronounced in ethanol. The presence of multiple emitting species has been confirmed by lifetime measurements. | ||
| Fig. 9 Absorption (···), emission (———, right; λex = 358 nm) and emission excitation (———, left; λem = 421 nm) spectra of TRN in MTHF at 77 K. The intensity of the excitation spectrum has been normalized to the 370 nm band of the absorption spectrum. | ||
 | ||
| Fig. 10 Absorption (···), emission (———–, right; λex = 350 nm) and emission excitation (———, left; λem = 430 nm) spectra of TRN in MTHF solution at room temperature. The intensities of the emission and emission excitation spectra have been normalized to the 370nm band of the absorption spectrum. | ||
The room temperature spectra are similar to those previously published for TRN in (impure) cyclohexane18 and reproduced by us using 3MP which had not been rigorously purified. We attribute the additional weak absorption band with an onset near 400 nm in MTHF and ethanol and a majority of the emission at room temperature in both solvents to trace amounts of highly fluorescent moieties of undetermined structure but possibly of photochemical origin. We also suggest that the presence of multiple emitting species is almost certainly responsible for many of the large disparities in the spectra, quantum yields and fluorescence lifetimes previously reported for TRN in fluid organic solvents at room temperature.16–19
Conclusions
The first systematic study of the spectroscopy and photophysics of tropolone in condensed media has been undertaken; the important variables of solvent and solute purity, solvent structure, temperature and excitation wavelength have been controlled. The results are consistent with more detailed spectroscopic7,10–13 and recent dynamic14 measurements of tropolone and its van der Waals complexes and clusters in supersonic expansions.In weakly interacting solvents, such as 3-methylpentane, the only species yielding significant fluorescence is the S1, 1(π,π*) state of the intramolecularly hydrogen-bonded neutral molecule. In acidic aqueous solution at pH<3, the emitting state is also 1(π,π*) S1, but the intramolecular hydrogen bond has been disrupted by the solvent. In basic aqueous solution at pH>8 the emitting species is the corresponding excited anion. In many other solvents, impurity emission makes reliable measurements difficult at room temperature. Contrary to previous reports, no emission from solvent-stabilised 1(n,π*) excited states is observed.
In glass-forming media the fluorescence quantum yields and excited state lifetimes of the major emitting species increase in a sigmoidal fashion as the temperature decreases, and are a factor of more than 100 greater at 77 K than at room temperature. The same effect is observed in the intramolecularly hydrogen-bonded neutral molecule, the water solvated neutral molecule and the anion. The fluorescence quantum yields also decrease with increasing excitation energy in the UV, an effect which is most pronounced in more weakly interacting solvents at temperatures near room temperature.
A mechanism which rationalises the observed effects has been proposed. Both the spectra and the excited state temporal decays indicate that emission from short-lived, vibrationally unrelaxed S1 constitutes the majority of the fluorescence at room temperature, a small fraction of the total e mission at 77 K, and an increasing fraction of the total emission with decreasing excitation wavelength. The shapes of ϕem(T) and τ(T) have been attributed to the effects of temperature on the dynamics of solvent relaxation based on qualitative correlations between the observed parameters describing the effects in different media and known characteristics of the media, including their glass transition temperatures and polarity.
Acknowledgements
The authors gratefully acknowledge the continuing support of this research by the Natural Sciences and Engineering Research Council of Canada. The authors are pleased to acknowledge the contributions of Dr YouXian Wen to the group's initial work on intramolecular excited state proton transfer.References
- K. P. Ghiggino, A. D. Scully and S. W. Bigger, ACS Symp. Ser., 1988, 381, 57 Search PubMed.
- G. A. Kraus, W. Zhang, M. J. Fehr, J. W. Petrich, Y. Wannemuehler and S. Carpenter, Chem. Rev., 1996, 96, 523 CrossRef CAS.
- D. S. English, K. Das, J. M. Zenner, W. Zhang, G. A. Kraus, R. C. Larock and J. W. Petrich, J. Phys. Chem. A, 1997, 101, 3235 CrossRef CAS.
- P. Chou, D. McMorrow, T. J. Aartsma and M. Kasha, J. Phys. Chem., 1984, 88, 4596 CrossRef CAS.
- P. Ball, Made to Measure: New Materials for the 21st Century, Princeton University Press, Princeton, NJ, 1996. Search PubMed.
- T. Nishiya, S. Yamauchi, N. Hirota, M. Baba and I. Hanazaki, J. Phys. Chem., 1986, 90, 5730 CrossRef CAS.
- For a review, see, V. J. MacKenzie and R. P. Steer, Res. Chem. Intermed., 1998, 24, 813. Search PubMed.
- D. J. Jang and D. F. Kelley, J. Phys. Chem., 1985, 89, 209 CrossRef CAS.
- M. Jinguji, M. Ishihara, T. Nakazawa, T. Hikida and Y. Mori, J. Photochem. Photobiol., A, 1992, 66, 33 Search PubMed.
- A. C. P. Alves and J. M. Hollas, Mol. Phys., 1972, 23, 927 CAS.
- H. Hamabe, T. Fukuchi, S. Shiraishi, K. Nishi, Y. Nishimura, T. Tsuji, N. Nishi and H. Sekiya, J. Phys. Chem. A, 1998, 102, 3880, and references therein. Search PubMed.
- R. K. Fr ost, F. C. Hagemeister, C. A. Arrington, D. Scheppenbach, T. S. Zwier and D. Jordan, J. Chem. Phys., 1996, 105, 2605 CrossRef CAS.
- A. Mitsuzuka, A. Fujii, T. Ebata and N. Mikama, J. Chem. Phys., 1996, 105, 2618 CrossRef CAS.
- V. J. MacKenzie, H. K. Sinha, S. C. Wallace and R. P. Steer, Chem. Phys. Lett., 1999, 305, 1 CrossRef CAS.
- For a review, see, E. C. Lim, Adv. Photochem., 1997, 23, 165. Search PubMed.
- R. Croteau and R. M. Leblanc, Photochem. Photobiol., 1978, 28, 33 Search PubMed.
- R. Croteau and R. M. Leblanc, J. Lumin., 1977, 15, 353 CrossRef CAS.
- E. F. Bréhéret and M. M. Martin, J. Lumin., 1978, 17, 49 CrossRef CAS.
- B. Bhattacharyya and J. Wolff, Proc. Natl. Acad. Sci, U.S.A., 1974, 71, 2627 CAS.
- P. Chieux, NATO ASI Ser., Ser. C, 1989, 205, 359 Search PubMed.
- D. R. James, D. R. M. Demmer, R. E. Verrall and R. P. Steer, Rev. Sci. Instrum., 1984, 54, 1121 CrossRef CAS.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229 CrossRef CAS.
- S. R. Meech and D. Phillips, J. Photochem., 1983, 23, 193 Search PubMed.
- R. Richert and A. Wagener, J. Phys. Chem., 1991, 95, 10115 CrossRef CAS.
- C. A. Angel and E. J. Sare, J. Chem. Phys., 1970, 52, 1058 CrossRef.
- S. K. Brahma, W. F. Howard, Jr. and W. H. Nelson, J. Phys. Chem., 1984, 88, 5551 CrossRef CAS.
- J. Okubo, F. Imaizumi, T. Nomura, T. Hoshi and M. Kobayashi, Nippon Kagaku Kaishi, 1989, 10, 1733 Search PubMed.
- B. Dick, J. Phys. Chem., 1990, 94, 5752 CrossRef CAS.
- T. Förster, Z. Elektrochem., 1950, 54, 42 Search PubMed.
- M. Maroncelli, J. MacInnis and G. R. Fleming, Science, 1989, 243, 1674, and references therein. Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, VCH, Weinheim, 1988. Search PubMed.
- S. P. McGlynn, T. Azumi and M. Kinoshita, Molecular Spectroscopy of the Triplet State, Prentice-Hall, Englewood Cliffs, NJ, 1969. Search PubMed.
| This journal is © the Owner Societies 2000 |