Oligoacetylenic sulfides
Albert W. M.
Lee
*,
Anissa B. W.
Yeung
,
Mabel S. M.
Yuen
,
H.
Zhang
,
X.
Zhao
and
W. Y.
Wong
Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Kowloon, Hong Kong.. E-mail: alee@hkbu.edu.hk
First published on 7th January 2000
Abstract
Linear acetylenic and diacetylenic sulfides consisting of up to eight triple-bond units with alternating sulfur atoms and acetylene units were synthesized.
The synthesis of conjugated or homoconjugated cyclic or linear oligoacetylene systems is a rapidly developing research frontier and has attracted considerable attention from both fundamental and applied viewpoints.1 These all-carbon or carbon-rich acetylene-based scaffolds are expected to exhibit a variety of unusual structural, electronic, electrical and optical properties.2–4 Linear rigid oligoacetylenic molecular rods of defined length in nanometer-sized structures could also serve as molecular wires in molecular electronic application.5–7
For the conjugated oligoacetylenic systems, a representative example is a compound with six conjugated diacetylene units synthesized by Diederich’s group through end-capping polymerization.8 Molecular wires end-capped with redox-active metal groups have also been reported.9,10 The longest molecule of this group has up to ten conjugated acetylene units. Novel heterocycles comprising alternating phosphorus atoms and acetylene units reported by Scott’s group are also known.11 Dendrimers with acetylenic units12 and alkynyl sulfides13 have also been prepared. However, to the best of our knowledge, there is no report on linear oligoacetylenic compounds containing heteroatom bridging between all or some of the acetylene units. Based on our experience in the preparation and uses of acetylenic sulfoxides and related compounds in organic synthesis,14 a systematic approach to the syntheses of oligomeric acetylenic sulfides with up to eight acetylene units is reported here.
Two key reactions, namely sulfurization and oxidative coupling via Hay and Glaser-type related methods,15 were used in building up the oligomeric acetylenic sulfides. Chain length is doubled in each cycle of the reaction.
The crucial starting material, a mono-protected bis-acetylene sulfide 7, was first prepared. When an equal molar ratio of trimethylsilylacetylene 1 and triisopropylsilylacetylene 2 was treated with 1 equiv. of BuLi followed by 0.5 equiv. of SCl2 at −78 °C, a mixture of silylated bis-acetylene sulfides (3–5) were formed (Scheme 1). The reaction mixture was placed under mild desilylation conditions (K2CO3/MeOH). The TMS group was hydrolyzed while the TIPS group remained intact. The volatile bis-acetylene sulfide 6 resulting from the hydrolysis of 3 was lost during work up and solvent evaporation. The remaining mono- and di-TIPS bis-acetylene sulfides 7 and 4 could be easily separated by column chromatography, with the mono-silylated compound 7 being the more polar component. The overall isolated yield of the desirable mono-protected compound 7 was 40%. The recovered di-TIPS acetylene sulfide 4 could also serve as the precursor of 7 through careful monodesilylation with KF in MeOH under close monitoring with TLC.
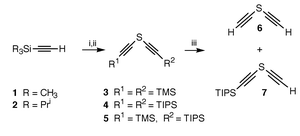 | ||
| Scheme 1 Reagents and conditions: i, BuLi; ii, SCl2, −78 °C; iii, K2CO3, MeOH. | ||
Oxidative coupling and sulfurization reactions were then used in the chain elongation of the acetylenic sulfide. Several sets of oxidative coupling conditions for acetylene compounds were studied. Eventually, we found that the original Hay conditions16 using CuCl and TMEDA afforded satisfactory results. For sulfurization, freshly distilled SCl2 was used. Alternatively, a stable solid sulfurization agent, bis(benzenesulfonyl) sulfide 8,17 used extensively by Scott could also be used. As shown in Scheme 2, oxidative coupling of 7 afforded diacetylene compound 9 in 94% yield. Sulfurization of 7 with SCl2 afforded sulfide 10 in 72% yield. Mono-deprotection of 10 to 12 was achieved by a carefully controlled desilylation procedure using KF in THF–H2O in the presence of a catalytic amount of Bu4NBr. The reaction was monitored closely via TLC and was stopped when 11 started to appear. Mono-deprotected 12 could be isolated in 35% yield after column chromatography with the recovery of about 16% of the starting material. If the desilylation reaction was allowed to run for a longer time at room temperature, the unprotected terminal acetylene compound 11 could also be isolated. The unprotected and mono-protected acetylenic sulfides 11 and 12 are very unstable, especially when concentrated.
 | ||
| Scheme 2 Reagents and conditions: i, CuCl, TMEDA, air, CHCl3; ii, BuLi or LIHMDS, then SCl2 or 8 (PhSO2SSO2Ph); iii, KF, THF–H2O, Bu4NBr (cat.). | ||
Freshly prepared 12 was subjected to the coupling and sulfurization cycles to extend the chain to eight acetylene units. Coupling afforded diacetylene compound 13 in 55% yield. For the sulfurization process, we found that BuLi reacted with the triple bonds of 12 to give complicated products. Therefore, LIHMDS was used instead and 14 was prepared in 25% yield using 8 as the sulfurization agent. Monodesilylation of 9 to 15 could also be achieved under carefully controlled conditions. However, the coupling product 16, which could be detected by FAB MS, was extremely unstable and could not be isolated in pure form.
All these new acetylenic sulfides were characterized by NMR, IR and MS. Some of the spectral data are summarized in Table 1. The typical IR frequency of the triple bond is around 2100 cm−1, and the terminal acetylene C–H signal appears around 3300 cm−1. UV spectra of oligoacetylene sulfides 4, 10, 13 and 14 were also recorded. There is no observable bathochromic shift (λmax = 231–237 nm) for compounds 4, 10 and 14 as the chain length increased. This indicated that the degree of conjugation between the acetylene units is weak. Compound 13, being a diacetylene compound, showed a slight bathochromic shift (λmax = 260 nm) relative to compounds 4, 10, 14 and had a large molar absorptivity.
| δC | |||||
|---|---|---|---|---|---|
| Compound | sp3 (Pri) | sp | δH | νmax/cm−1 | λmaxa/nm (ε/dm3 mol−1 cm−1) |
a
In cyclohexane.
b
C![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) CH. CH.
|
|||||
| 4 | 11.23, 18.49 | 87.75, 100.49 | 1.05(s) | 2098 | 231 (20600) |
| 7 | 11.23, 18.49 | 67.82, 84.22,b 86.18, 101.29 | 1.06(s), 2.97(s) | 3303, 2103 | |
| 9 | 11.23, 18.51 | 68.50, 82.09, 84.48, 102.84 | 1.11(s) | 2104 | |
| 10 | 11.22, 18.51 | 81.56, 83.06, 85.97, 101.37 | 1.07(s) | 2102 | 231 (41500) |
| 11 | 66.38, 81.94, 82.16, 84.66b | 2.99(s) | 3289, 2098 | ||
| 12 | 11.20, 18.51 | 66.51, 81.21, 81.65, 82.53, 83.58, 84.58b, 85.77, 101.47 | 1.07(s), 2.99(s) | 3297, 2103 | |
| 13 | 11.23, 18.53 | 67.45, 79.87, 80.66, 81.69,83.85, 83.92, 85.68, 101.65 | 1.12(s) | 2101 | 260 (97400) |
| 14 | 11.22, 18.53 | 80.85, 81.26, 81.74, 82.01,82.55, 83.67, 85.72, 101.58 | 1.07(s) | 2100 | 237 (63900) |
We also explored the chemistry of double conjugate addition of nucleophiles to the terminal positions of the bis-acetylene sulfides and the corresponding sulfones.18 Reactions with Na2S impregnated on neutral alumina19 took place readily in DMF–MeOH mixture at 0 °C and room temperature respectively for 4 and 17 (Scheme 3). However, to our surprise the monodesilylated compounds 18a,b were the only isolated products. The structure of cyclic sulfone 18b was confirmed by X-ray analysis (Fig. 1).†
 | ||
| Scheme 3 Reagents and conditions: i, Na2S, Al2O1, DMF–MeOH. | ||
 | ||
| Fig. 1 X-Ray structure of 18b. | ||
In summary, the first syntheses of a series of oligoacetylenic sulfides with alternating sulfur atoms and acetylene or diacetylene units were accomplished using the monoprotected bis-acetylene sulfide 7 as the starting material. Studies on the synthesis of some cyclic and metal end-capped analogs20 are in progress.
Acknowledgements
Financial support from the Research Grants Council (RGC/97-98/48, HKBU 2048/97P) is gratefully acknowledged.References
- P. J. Stang and F. Diederich, Modern Acetylene Chemistry, VCH, Weihheim, 1995. Search PubMed.
- R. Gleiter, Angew. Chem., Int. Ed. Engl., 1992, 31, 27 CrossRef.
- D. Bloor, Chem. Br., 1995, 385 Search PubMed.
- F. Diederich, Nature, 1994, 369, 199 CrossRef CAS.
- J. M. Tour, Chem. Rev., 1996, 96, 537 CrossRef CAS.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89 CrossRef.
- M. D. Ward, Chem. Br., 1996, 568 Search PubMed.
- J. Anthony, C. Boudon, F. Diederich, J.-P. Gisselbrecht, V. Gramlich, M. Gross, M. Hobi and P. Seiler, Angew. Chem., Int. Ed. Engl., 1994, 33, 763 CrossRef.
- W. Weng, T. Bartik, M. Brady, B. Bartik, J. A. Ramsden, A. M. Arif and J. A. Gladysz, J. Am. Chem. Soc., 1995, 117, 11922 CrossRef CAS.
- N. L. Narvor, L. Toupet and C. Lapinte, J. Am. Chem. Soc., 1995, 117, 7129 CrossRef.
- L. T. Scott and M. Unno, J. Am. Chem. Soc., 1990, 112, 7823 CrossRef CAS.
- Z. Xu and J. J. Moore, Angew. Chem., Int. Ed. Engl., 1993, 32, 246 CrossRef; 1993, 32, 1354..
- G. Lábbée, B. Haelterman and W. Dehaen, J. Chem. Soc., Perkin Trans. 1, 1994, 2203 RSC.
- A. W. M. Lee and W. H. Chan, Top. Curr. Chem., 1997, 190, 103 Search PubMed.
- G. Eglinton and W. McCrae, in Advances in Organic Chemistry: Methods and Results, ed. R. A. Raphael, E. C. Taylor and H. Wynberg, Interscience, New York, 1963. Search PubMed.
- A. S. Hay, J. Org. Chem., 1962, 27, 3320 CrossRef CAS.
- F. DeJong and M. J. Janssen, J. Org. Chem., 1971, 36, 1645 CrossRef.
- J. Meijer, P. Vermeer, H. D. Verkruijsse and L. Brandsma, Recl. Trav. Chim. Pays-Bas., 1973, 92, 1326 Search PubMed.
- B. Czech, S. Quici and S. L. Regen, Synthesis, 1980, 113 CrossRef CAS.
- S. L. James, M. Younus, P. R. Raithby and J. Lewis, J. Organomet. Chem., 1997, 543, 233 CrossRef CAS.
Footnote |
† Crystal data for
18b: C13H24O2S2Si,
M = 304.53, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a =
7.9504(5), b = 15.857(1), c = 16.001(1) Å, α
= 118.632(1), β= 98.449(1), γ = 97.355(1)°, V =
1705.0(2) Å3, Z = 4, T = 293 K,
μ(Mo-Kα) = 3.76 cm−1, 10061 reflections measured,
7271 unique, R(int) = 0.0128, final R1 =
0.039, wR2 = 0.1061 (based on
F2) for 7271 [I >
2ς(I)] observed reflections. CCDC 182/1493. See
http://www.rsc.org/suppdata/cc/a9/a908220d/ for crystallographic data in
.cif format. (no. 2), a =
7.9504(5), b = 15.857(1), c = 16.001(1) Å, α
= 118.632(1), β= 98.449(1), γ = 97.355(1)°, V =
1705.0(2) Å3, Z = 4, T = 293 K,
μ(Mo-Kα) = 3.76 cm−1, 10061 reflections measured,
7271 unique, R(int) = 0.0128, final R1 =
0.039, wR2 = 0.1061 (based on
F2) for 7271 [I >
2ς(I)] observed reflections. CCDC 182/1493. See
http://www.rsc.org/suppdata/cc/a9/a908220d/ for crystallographic data in
.cif format. |
| This journal is © The Royal Society of Chemistry 2000 |