An infrared reflectance spectroscopic study of a pH-tunable network of nanoparticles linked by hydrogen bonding
Wenxia Zheng, Mathew M. Maye, Frank L. Leibowitz and Chuan-Jian Zhong*
Department of Chemistry, State University of New York at Binghamton, Binghamton, New York 13902, USA.. E-mail: cjzhong@binghamton.edu
First published on UnassignedUnassigned7th January 2000
Abstract
This paper presents novel findings of the pH-tunable structural properties of a hydrogen-bonded nanoparticle network using an infrared reflectance spectroscopic (IRS) technique. Gold nanoparticles encapsulated with thiolates and alkylthiols terminated with carboxylic groups were utilized as building blocks and molecular linkers, respectively, for assembling the network film. The IRS bands associated with the reactivities of the carboxylic acid groups on the nanoparticles allowed us to analyze the structures during surface titration of the network thin films. IRS data have revealed that the network, linked by predominantly head-to-head hydrogen bonding, could be reversibly tuned between the neutral protonated and the ionic deprotonated states. This protonation/deprotonation process was also found to exhibit membrane-type properties, as supported by open-circuit potential measurement of the membrane. These findings have important implications to designing nanoparticle-based functional nanostructures for molecular recognition.
Introduction
Metallic or semiconductive nanoparticles encapsulated with alkanethiolates are interesting because there are many potential technological applications, including microelectronics, optical devices, magnetic materials, catalysis and chemical sensing.1–4 An area rapidly emerging from nanochemistry and nanomaterials fields involves the exploration of utilities of the molecular shell and the nanoscale core as building blocks towards nanostructured functional materials. Nanoconstruction based on functionalized thiol linking strategies has recently been demonstrated for metal and semiconductor nanoparticles via stepwise layer-by-layer pathway,5–7 DNA-based method,6b and an exchanging–crosslinking–precipitation route.8 While exhibiting interesting electrical, electrochemical and optical properties, most of the nanostructures do not allow subsequent structural tuning because of the nature of the covalent linkages. In view of the immense opportunities of nanostructural tuning in chemical and biological applications (e.g., molecular recognition and drug delivery),9 here we report non-covalent linkages in nanoconstruction of interfacial tunable network films. Gold nanoparticles encapsulated with thiolates terminated with carboxylic acid (–COOH) were chosen as a model system based on the recent abundant demonstration for the synthesis and characterization.1 We have demonstrated for the first time that the chemical reactivity at this core–shell combination leads to networks of head-to-head hydrogen-bonding linkages at the encapsulating molecular shells. The network can be reversibly tuned by pH in between neutral and ionic membrane-type architectures.While there has recently been considerable interest in the fabrication of core–shell nanoparticles with a variety of functionalized shell encapsulations,1 including recently thiols terminated with carboxylic acid,10,11 the utilization of non-covalent linkages, such as hydrogen bonding, for constructing supramolecular network with tunable properties is not demonstrated. Hydrogen-bonding phenomena exist in nature, such as base-pairing of nucleic acids and helix–coil transition of DNA. The understanding of hydrogen bonding as tunable linkages in nanoparticle networks has thus important implications to many fundamental issues in emerging fields, including molecular engineering of 3D nanostructured materials1,9 and 2D monolayers.12,13 We have recently demonstrated a straightforward ‘exchanging–crosslinking–precipitation’ route for the preparation of dithiol-linked gold nanoparticle films.8 We now show that this route can be utilized to prepare the hydrogen-bonded network of gold nanoparticles, and the use of an infrared reflectance spectroscopic (IRS) technique allows us to analyze the detailed nanostructured properties in the network.
Experimental
The main chemicals used included decanthiol (DT, 96%), 11-mercaptoundecanoic acid (MUA, 97%), hydrogen tetrachloroaurate (HAuCl4, 99%), tetraoctylammonium bromide (TOABr, 99%), sodium borohydride (NaBH4, 99%). All chemicals (from Aldrich, Milwaukee, WI 53201, USA) were used as received. Water was purified with a Millipore Milli-Q water system (Bedford, MA 01730, USA).The preparation of 5 nm core sized Au (Au5 nm) nanoparticles encapsulated with DT was described recently,14 which involves a two-phase protocol to synthesize 2 nm core sized particles15 followed by heating-induced size evolution.14 The preparation of the thin films was via a one-step ‘exchanging–crosslinking–precipitation’ route, which was detailed in our recent publication.8 Briefly, the nanoparticles and MUA were first mixed in a toluene solvent with a controlled ratio (e.g., [MUA]/[Au5 nm] ≈ 400, at 0.5 μM Au5 nm and 0.2 mM MUA). Substrates of evaported gold film slides or polycrystalline gold, after surface pre-treatments, were immersed into the solution. The substrates were pre-cleaned with 1∶3 H2O2 (30%)–H2SO4 (conc.) solution and thoroughly rinsed with deionized water. The gold film surfaces were pre-coated with an octyldecanethiolate monolayer to enhance adhesion of the film. This adlayer effectively enhances the adhesion because of its strong hydrophobic interaction, though a detailed mechanism is still under investigation. The nanoparticle network film thickness was monitored by both UV–VIS absorbance of the surface plasma band and the mass-loading measured using a quartz-crystal microbalance (QCM).8
IRS spectra were acquired with a Nicolet (Madison, MI 53711, USA) 760 FT-IR spectrometer equipped with a liquid N2-cooled HgCdTe detector and a variable-angle reflectance device. The incident beam was p-polarized light with a grazing angle of 82° with respect to the surface normal. A gold slide coated with octadecanethiolate-d37 monolayer was used as the IRS reference. The curve fitting applied to the deconvolution was based on the convergence of a Marquardt–Levenberg algorithm to values that gave the best least-squares fit to the experimental data. The peak positions, absorbances, widths and areas were determined by deconvoluting the overlapping curves with a Lorentzian-type band shape maintaining constant width for each peak from different pH conditions.
The open-circuit potential (OCP) measurement was performed on an EG&G (Princeton, NJ 08543, USA) Model 273A potentiostat (input impedance ≈1012 Ω), using a Ag/AgCl (saturated KCl) electrode as the reference. The solution was thoroughly purged with argon before measurements. The pH was adjusted by adding 0.11 M NaOH into a 0.1 M KClO4 + 0.01 M H3PO4 solution (constant ionic strength12).
Results and discussion
The decanethiolate-encapsulated Au5 nm and COOH-terminated alkyl thiols were used as the networking precursors. Similar to the dithiol-linked nanoparticle films we recently reported,8 the formation of the network films involved an exchange of the thiols with the gold-bound thiolates followed by crosslinking and precipitation via hydrogen bonding at the terminals. While we have studied COOH-terminated thiols of several chain lengths, this report focuses on the results from the 11-mercaptoundecanoic acid (MUA) system. The thickness of the precipitated thin films could be controlled by the immersion time, while the absorbance of the nanoparticle surface plasmon band and the mass-loading were monitored by UV–VIS and QCM methods. The films were stable and displayed uniform colors ranging from purple to green depending on their thickness. The films were insoluble in toluene or hexane, but soluble in ethanol. The optical and morphological properties in the MUA–Au5 nm films were characterized by the surface plasmon resonance band (570–600 nm) and the supramolecular-type aggregate features in transmission electron microscopic data.Fig. 1 presents the IRS data in the
C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O stretching region of –COOH as the direct evidence for the
interparticle hydrogen-bonding linkages in the MUA–Au5 nm
film (A). The spectrum for a 2D MUA monolayer (B) is also included for
comparison. Note that spectrum A has an overall absorbance of ≈50 times
larger than spectrum B, a fact presumably due to a combination of the
nanoparticle multilayer (10–20 equivalent) and surface
enhancement16 effects. While both spectra
exhibit an envelope of three band components as represented by the dashed
curves derived from spectral deconvolution, spectrum A shows remarkable
distinctions from spectrum B in terms of the band position and relative
weight. For the MUA monolayer, these bands were previously assigned17 to ν(CO)COOH in free acid
or non-hydrogen bonded (1, 1741 cm−1), side-by-side
dimeric hydrogen bonded (2, 1718 cm−1), and polymeric
hydrogen bonded (3, ≈1690 cm−1) states. In contrast,
bands of the hydrogen-bonded modes for the MUA–Au5 nm
network film are shifted to lower energies, i.e., 1710
cm−1 (2) and ≈1670 cm−1 (3). The free
acid band (1) remains basically unchanged (1740 cm−1 (1)).
These shifts in ν(CO)COOH are remarkable, and can be
attributed to the formation of the head-to-head hydrogen bonding in the
nanostructure. In fact, the wavenumber of band 2 is identical to the band
observed for the cis-configured head-to-head hydrogen-bonded dimer
in the condensed phases of alkanoic acids.18 A contribution from
the trans-configuration of the hydrogen-bonded dimer18a to the broad band 3, in addition to
polymeric hydrogen bonding, may also be operative. These binding modes are
impossible in the 2D MUA monolayers. Moreover, while the 2D monolayer
exhibits ≈40% free acid component (1, B), the MUA–Au5
nm film shows only a level of <10%. The results thus provide clear
evidence for the predominant head-to-head hydrogen-bonded linkage in the
network.
O stretching region of –COOH as the direct evidence for the
interparticle hydrogen-bonding linkages in the MUA–Au5 nm
film (A). The spectrum for a 2D MUA monolayer (B) is also included for
comparison. Note that spectrum A has an overall absorbance of ≈50 times
larger than spectrum B, a fact presumably due to a combination of the
nanoparticle multilayer (10–20 equivalent) and surface
enhancement16 effects. While both spectra
exhibit an envelope of three band components as represented by the dashed
curves derived from spectral deconvolution, spectrum A shows remarkable
distinctions from spectrum B in terms of the band position and relative
weight. For the MUA monolayer, these bands were previously assigned17 to ν(CO)COOH in free acid
or non-hydrogen bonded (1, 1741 cm−1), side-by-side
dimeric hydrogen bonded (2, 1718 cm−1), and polymeric
hydrogen bonded (3, ≈1690 cm−1) states. In contrast,
bands of the hydrogen-bonded modes for the MUA–Au5 nm
network film are shifted to lower energies, i.e., 1710
cm−1 (2) and ≈1670 cm−1 (3). The free
acid band (1) remains basically unchanged (1740 cm−1 (1)).
These shifts in ν(CO)COOH are remarkable, and can be
attributed to the formation of the head-to-head hydrogen bonding in the
nanostructure. In fact, the wavenumber of band 2 is identical to the band
observed for the cis-configured head-to-head hydrogen-bonded dimer
in the condensed phases of alkanoic acids.18 A contribution from
the trans-configuration of the hydrogen-bonded dimer18a to the broad band 3, in addition to
polymeric hydrogen bonding, may also be operative. These binding modes are
impossible in the 2D MUA monolayers. Moreover, while the 2D monolayer
exhibits ≈40% free acid component (1, B), the MUA–Au5
nm film shows only a level of <10%. The results thus provide clear
evidence for the predominant head-to-head hydrogen-bonded linkage in the
network.
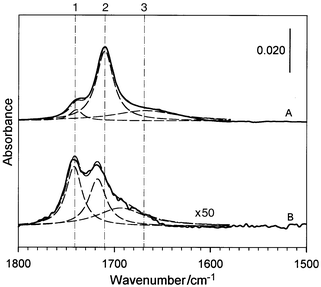 | ||
| Fig. 1 The IRS spectra for a MUA–Au5 nm network film (A), and a MUA monolayer (B). The dashed lines represent spectral deconvolution based on Lorentzian-type envelope. | ||
Two additional IRS spectral features revealed further supporting evidence for the network structure. First, the methylene stretching band positions (2918 and 2848 cm−1) in the 3D film are only slightly different from those in the MUA monolayer (2920 and 2850 cm−1), indicative of largely comparable chain–chain packing. Secondly, the nanoparticle film showed a distinctive absorbance rising from ≈1000 cm−1 to near infrared due to surface plasmon resonance in a continuous conductive film.6a,19 This was also evidenced by preliminary data of the conductivity dependence on the core size and the linkage shell chain length.
In Fig. 2, we demonstrate the pH-tuning
properties of the nanostructured network. The spectra, taken upon immersion
in different pH solutions (constant ionic strength), are shown in the
diagnostic region of –COOH (ν(CO)) and
CO2− (ν(C–O)) stretching vibrations
(Fig. 2A). As the pH increases (from a to
f), the ν(CO) bands display a gradual decrease in intensity and
disappear at pH ≈ 10, whereas new bands corresponding to the asymmetric
and symmetric ν(C–O) bands of
–CO2−,17b–di.e., 1578
cm−1 (νa) and ≈1435 cm−1
(νs), emerge at pH > 6. In pH 6–10, the detection of
both –COOH and –CO2− species is
indicative of partial protonation/deprotonation. Remarkably, the pH
dependence is reversible, evidenced by the reappearance of the
ν(CO) bands after a low pH treatment (g). The detectable ≈10%
decrease in absorbance is probably in part due to a structural
reorganization, which we believe is small based on the insignificant
changes of the C–H stretching bands. The above
protonation–deprotonation reactivity at the carboxylic linkage of the
hydrogen-bonded nanoparticle network is illustrated in Scheme 1. It is important to point out that the
observation of this reversible chemistry at the linkages is intriguing
because the network thin film remains intact. This has implications in
developing this network film in molecular recognition applications
involving the reversible pH-tunable structural properties.
| ||
| Fig. 2 (A) The pH dependence of IRS spectra of the MUA–Au5 nm network film immersed in solutions of pH values of 2.13 (a), 4.02 (b), 5.99 (c), 7.96 (d), 10.18 (e), 11.80 (f) and 2.32 (g). (B) Plots of the peak areas for IRS bands 1, 2, 3, and 4, as well as the sum of the first three peaks (1 + 2 + 3), as a function of the pH. These peak areas are derived based on spectral deconvolution. The insert represents the pH dependence of the open-circuit potential. | ||
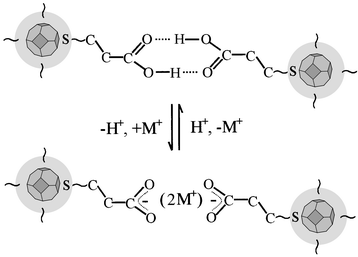 | ||
| Scheme 1 A schematic illustration of the protonation–deprotonation reactivity at the head-to-head hydrogen-bonding based linkage in the nanoparticle network (only a fraction of the network is illustrated). | ||
We further questioned whether there is any difference in the pH tuning for the various hydrogen-bonded and non-hydrogen-bonded –COOHs. In an effort to answer this question, the peak area of each band and the total as well are plotted against pH (Fig. 2B). Note that only the va band (4) is plotted for the CO2− because the νs band (5) is overlapped with several bands including δ(CH2, ≈1415 cm−1) and ν(C–O) + δ(OH) (1430–1460 cm−1). As is evident, while band 2 decreases gradually in the low pH range, bands 1 and 3 do not show a decrease until pH 5–6. This result suggests that the pH tuning is most effective for the head-to-head hydrogen-bonded linkage. Furthermore, the pH 6–10 range between the disappearance of the ν(COOH) bands and the onset of the νa(CO2−) bands with a cross-point of the total at pH ≈8 may reflect an approximate interfacial pKa range of the overall protonation/deprotonation in the nanostructured film. Precise determination will need to consider many other factors including the solution concentrations.12,13,20
The result may be indicative of the possible existence of membrane-type properties21 in the nanostructured film, which is further examined by measuring the OCP during the pH tuning. OCP is the electrode potential across the entire network film interfaces at which there is no net charge transfer. A change in the OCP reflects a perturbation to the electrical double layer, which includes charge transfer across the interface (e.g., redox reaction equilibrium), ionic charges in the film (e.g., ion-exchanger membrane potential), as well as effects due to surface adsorption and dipole reorientation. Since the ionic charges are associated with the ionized fraction of the acid moieties, they contribute to the potential distribution across the interfaces.12,13 OCP thus provides a measure of the interfacial membrane potential as a result of the change between the neutral –COOH and the charged –CO2− network. A representative set of the preliminary OCP data is included in Fig. 2B for comparison. The decrease of the OCP with increasing pH (in degassed solutions with a constant ionic strength) is qualitatively consistent with the membrane potential developed across the increasingly negatively charged network.21 The small but notable transition at pH 6 corresponds approximately to the onset of deprotonation detected by the IRS. Interestingly, this pH value, while smaller by 2–4 units than most COOH-terminated monolayer counterparts, is comparable with the pKa values for alkanoic acid in different solutions.20 This result likely reflects the membrane-type protonation/deprotonation properties of the network film.19b,c We further note an additional hint of transition at pH ≈10, which is observable in at least five sets of OCP data. Whether these transitions reflect a difference in two protonation sites (e.g., one hydrogen-bonded linkage and the other free acid), or a difference in the effective local proton concentration (the hydrophilic interparticle linkage and the less hydrophilic intraparticle environment), is under in-depth investigation. It is important to emphasize that the strucutral properties derived by the IRS surface titration of the interparticle linkages can not be directly translated by the structural properties of the solution counterparts, because of the effect of high surface packing density.
In summary, we have used the IRS technique to demonstrate for the first time that a thiolate-encapsulated gold nanoparticle network can be constructed via head-to-head hydrogen-bonded linkages of carboxylic shell components, and the network can be effectively tuned by pH to between neutral and ionic membrane-type properties. The network and reversible tuning serve as an intriguing example of chemical manipulation at interparticle non-covalent linkages of nanoparticle networks, and offers a new route to designing functional chemical sensing and molecular recognition elements. In addition, the IRS technique has been demonstrated to be particularly useful to unravel the binding structural details in the network films.
Acknowledgements
We thank Dr M. Porter for providing the initial gold/glass slides and IRS reference slides.References
- (a) M. J. Hostetler and R. W. Murray, J. Curr. Opin. Colloid Interface Sci., 1997, 2, 42 Search PubMed; (b) C. J. Kiely, J. Fink, M. Brust, D. Bethell and D. J. Schiffrin, Nature, 1998, 396, 444 CrossRef CAS.
- Nanoparticles and Nanostructured Films, ed. J. H. Fendler, Wiley-VCH, Weinheim, 1998. Search PubMed.
- M. Lahav, T. Gabriel, A. N. Shipway and I. Willner, J. Am. Chem. Soc., 1999, 121, 258 CrossRef CAS.
- C. R. Martin and D. T. Mitchell, Anal. Chem., 1998, 70, 322A CrossRef CAS.
- (a) M. Brust, D. Bethell, C. J. Kiely and D. J. Schiffrin, Langmuir, 1998, 14, 5425 CrossRef CAS; (b) M. Brust, D. Bethell, D. J. Schiffrin and C. J. Kiely, Adv. Mater., 1995, 7, 795 CAS; (c) M. Brust, D. Bethell, D. J. Schiffrin and C. J. Kiely, Adv. Mater., 1995, 7, 1655; (d) D. Bethell, M. Brust, D. J. Schiffrin and C. J. Kiely, J. Electroanal. Chem., 1996, 409, 137 CrossRef CAS.
- (a) M. D. Musick, D. J. Pena, S. L. Botsko, T. M. McEvoy, J. N. Richardson and M. J. Natan, Langmuir, 1999, 15, 844 CrossRef CAS; (b) C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS.
- K. Hu, M. Brust and A. J. Bard, Chem. Mater., 1998, 10, 1160 CrossRef CAS.
- (a) F. L. Leibowitz, W. X. Zheng, M. M. Maye and C. J. Zhong, Anal. Chem., 1999, 71, 5076 CrossRef CAS; (b) C. J. Zhong, W. X. Zheng and F. L. Leibowitz, Electrochem. Commun., 1999, 1, 72 CrossRef CAS.
- (a) M. J. Hostetler, C. J. Zhong, B. K. H. Yen, J. Anderegg, S. M. Gross, N. D. Evans, M. D. Porter and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 9396 CrossRef CAS; (b) A. C. Templeton, M. J. Hostetler, E. K. Wormoth, S. Chen, C. M. Hartshorn, V. M. Krishnamurthy, M. D. E. Forbes and R. M. Murray, J. Am. Chem. Soc., 1998, 120, 4845 CrossRef CAS; (c) Y. Liu, M. Zhao, D. E. Bergreiter and R. M. Crooks, J. Am. Chem. Soc., 1997, 119, 8720 CrossRef CAS; (d) M. Li, K. M. Wong and S. Mann, Chem. Mater., 1999, 11, 23 CrossRef CAS.
- S. R. Johnson, S. D. Evans and R. Brydson, Langmuir, 1998, 14, 6639 CrossRef CAS.
- C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763 CrossRef CAS.
- J. F. Smalley, K. Chalfant and S. W. Feldberg, J. Phys. Chem. B, 1999, 103, 1676 CrossRef CAS.
- (a) H. S. White, J. D. Peterson, Q. Cui and K. J. Stevenson, J. Phys. Chem. B, 1998, 102, 2930 CrossRef CAS; (b) C. P. Smith and H. S. White, Langmuir, 1993, 9, 1 CrossRef CAS; (c) M. A. Bryant and R. M. Crooks, Langmuir, 1993, 9, 385 CrossRef CAS; (d) K. Hu and A. J. Bard, Langmuir, 1997, 13, 5114 CrossRef CAS.
- (a) M. M. Maye, W. X. Zheng, F. L. Leibowitz, N. K. Ly and C. J. Zhong, Langmuir, in press; Search PubMed; (b) C. J. Zhong, W. X. Zheng, F. L. Leibowitz and H. H. Eichelberger, Chem. Commun., 1999, 13, 1211 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- C. W. Brown, L. Yue, J. A. Seelenbinder, P. Pivarnik, A. Rand, S. V. Letcher, O. J. Gregory and M. J. Platek, Anal. Chem., 1998, 70, 2991 CrossRef CAS.
- (a) R. G. Nuzzo, L. H. Dubois and D. L. Allara, J. Am. Chem. Soc., 1990, 112, 558 CrossRef CAS; (b) E. L. Smith, C. A. Alves, J. W. Anderegg, L. M. Siperko and M. D. Porter, Langmuir, 1992, 8, 2707 CrossRef CAS; (c) E. L. Smith and M. D. Porter, J. Phys. Chem., 1993, 97, 8032 CrossRef; (d) L. J. Kepley and R. M. Crooks, Anal. Chem., 1992, 64, 3191 CrossRef CAS; (e) Y.-T. Tao, W.-L. Lin, G. D. Hietpas and D. L. Allara, J. Phys. Chem. B, 1997, 101, 9732 CrossRef CAS.
- (a) S. Hayashi and J. Umemura, J. Chem. Phys., 1975, 63, 1732 CrossRef CAS; (b) J. Umemura, J. Chem. Phys., 1978, 68, 42 CrossRef CAS.
- F. Brouers, J. P. Clerc, G. Giraud, J. M. Laugier and Z. A. Randriamantany, Phys. Rev. B., 1993, 47, 666 CrossRef CAS.
- T. R. Lee, R. I. Carey, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1994, 10, 741 CrossRef CAS.
- (a) C. J. Zhong and K. Doblhofer, Electrochim. Acta, 1990, 35, 1971 CrossRef CAS; (b) K. Doblhofer, J. Electroanal. Chem., 1992, 331, 1015 CrossRef CAS; (c) A. Wojda and K. Maksymiuk, J. Electroanal. Chem., 1998, 441, 205 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |