Nanobiotechnology and its role in the development of new analytical devices†
Jean-Marc Lavala, Pierre-Emmanuel Mazeranb and Daniel Thomas*a
aLaboratoire de Technologie Enzymatique, UA6022 CNRS, University de Technologie de Compiègne, F-60205, Compiègne, France
bDépartement du Génie des Systèmes
Mécaniques, Université de Technologie de Compiègne, F-60205, Compiègne, France
First published on UnassignedUnassigned7th January 2000
Abstract
Physical methods of molecule observation and manipulation will prove useful, not only as research tools for investigating biomolecular structure and behavior, but also for the creation of nanostructures. Supramolecular and self-assembling structures are able to generate nanostructures, with many such systems being of biological origin. They form the interface between nanotechnology and biotechnology. Whereas biotechnological processes usually involve populations of cells or molecules, nanotechnological methods operate at the level of individual molecule manipulation. This article considers what advances have been made through cross-fertilisation between nanotechnology and biotechnology to develop for the next millennium new analytical tools at the microscale, using nanostructures as the sensitive part and with the ability to detect individual molecules.
The convergence of technologies being developed for machining materials at the atomic scale with technologies achieved for observing nanostructures has promoted the emergence and rapid growth of nanotechnology.
Supramolecular and self-assembling structures are able to generate nanostructures, many of such systems being of biological origin. They form the interface between nanotechnology and biotechnology.
Many biological systems—including viruses, membranes and protein complexes—are natural nanostructures. However, they may be analysed in the same way as non-biological structures, and their characteristics can be exploited in nanostructure design and development. It is becoming increasingly obvious that systems that integrate biological and chemical components with physical devices and electrodes have enormous potential for application, not only for developing analytical and monitoring devices, but also for molecular-scale bioreactors. Whereas biotechnological processes usually involve populations of cells or molecules (or, in some cases, individual cells or organelles), nanotechnological methods operate at the level of manipulation of individual molecules.
This article considers what advances have been made through cross-fertilisation between nanotechnology and biotechnology to develop: studies of single biological macromolecules (i.e., proteins); biotransducers in new analytical devices; and micro-total analysis systems.
A. Studies of single protein molecule
Since many decades ago, characterization of the functional and structural properties of biological molecules has been made by conventional techniques that operate on a population of biomolecules. Catalytic parameters of an enzyme determined by spectrophotometric measurement or the 3D structure of the enzyme obtained from X-ray diffraction are always the average behavior of an enzyme population. The current hypothesis is to assume that a homogeneous population of molecules allows the extrapolation of the results to the single molecule. In the last years, techniques appear to characterize and manipulate individual biomolecules, such techniques are more precisely scanning probes and optical techniques (see the recent reviews1–4). With these techniques, heterogeneity of populations of molecules has been identified.Scanning probe microscopy
Over these past years, scanning probe microscopy (SPM) and especially atomic force microscopy (AFM)5 has been intensively used to study biological samples at the nanometer scale. However, at the beginning, these techniques were not applied easily to biological specimens. Specific problems due to the technique were not known and some image interpretations were erroneous.6 Today the technique has matured and is reliable, the artifacts are well known and the image interpretation is generally not ambiguous. Concerning topographic imaging, some recent advances in sample preparations, operating conditions, probes and imaging mode have decreased the resolution to the nanometer level. For biological observations, the tip curvature radius (typically 10 nm to be comparable with the protein radius, i.e., albumin has a radius of 4 nm) and the protein compliance lead to great difficulties in observing the substructures of proteins adsorbed onto a solid surface. Nevertheless AFM has generated a growing interest due to the new capabilities it offers. It preserves the native state of molecules or cells because no sample preparations (fixation, dehydration, coatings, etc.) are needed and because proteins, e.g., bovine albumin,7 RNA polymerase8 or antibody9 can be explored in physiological media. Thus dynamic processes like antibody–antigen complex formation,10 enzyme activity11 or the DNA transcription12 have been observed. Another important aspect of AFM is its capability to measure the force between a sharp tip and the sample surface. This offers the possibility of measuring a wide range of properties from mechanical properties (elasticity, viscosity, plasticity, friction, etc.) to forces (attraction, adhesion, interaction, etc.) using various imaging modes. Nevertheless it is very difficult to measure quantitatively these properties. Generally, the signal of the imaging mode is just used to elaborate an image with an arbitrary unit opening the door to over-interpretation of data. For some imaging modes it is possible to realize a quantitative image of the tip/surface interaction (i.e., contact stiffness) but very difficult if not impossible to realize a quantitative image of the surface properties (i.e., elastic modulus).The atomic force microscope could be also used as a force-sensing instrument to measure the tip/surface force as a function of the distance and more interestingly the interaction energy (which is the integration of force with respect to distance).13 These two properties are important because they are responsible for dynamic behaviors like kinetic or affinity constants. Because the atomic force has been built for imaging it has several flaws concerning force-sensing measurement. The most notable of them is that the so-called force curve describes the tip/surface force as a function of the sample position instead of the tip/surface distance. One of the solutions to prevent this phenomena is to achieve an external force on the cantilever (via a magnet for example) to generate a true force tip/sample distance curve.14 But this has, to our knowledge, not been applied to biological experiment. Nevertheless this kind of experiment has been used to measure the avidin–biotin15–17 or antigen–antibody interactions18–20 or the elongation force of titin domains.21 Another interesting evolution of SPM is the development of the near field scanning optical microscope (SNOM).22–25 SNOM has been successfully used for biological investigations.26–30 Recent developments of probes25 and of shear force mode31 has decreased the resolution to a few nanometers.32 Future work could be the study of the luminescence activity of a single protein.
Optical techniques
Regrouping of Rhodamine molecules adsorbed on Si wafers was revealed as fluorescent spots with a measurable number of individual molecules.33 Fluorescence of the active site of an enzyme, i.e., FAD of cholesterol oxidase,34 allows one to follow the turnover of a single enzyme molecule. From the results of this kind of experiment, a new view of the population of enzyme molecules with a static and dynamic disorder of reaction rates appears. Individual enzyme molecules have different catalytic turnovers35 and a molecular memory phenomenon exists in the same enzyme molecule so that enzymatic turnovers are not independent of previous ones.34Recently,36 the rotation of F1 ATPase during ATP hydrolysis has been observed in an epifluoresence microscope. Visualization of the motion of the F1 subunit was effected by the attachment of a fluorescent actin to the F1 ATPase immobilized on a coverslip. Optical tweezers exploit the fact that light exerts force on matter. As in AFM, elasticity of macromolecules, i.e., titin,37 can be quantified by elongation or twisting of the molecule by optical tweezers. Optical tweezers also allow the measurement of the binding force between two biological molecules or movements. Applications concerned the linear motor proteins that move along a polymer track such as kinesin, myosin or RNA polymerase.4 Understanding of the mechanisms of the linear motor proteins allows the possibility of using these proteins as nanomotors for motions at the nanometer scale.
B. Biotransducers
Many technology applications in analysis require the organization of atoms or molecules in a two-dimensional space. Two approaches may be used to create the initial framework. First, as demonstrated by the microelectronic industry, such frameworks can be generated by the manufacture of silicon-based materials, which may then be machined with the desired structure or pattern. This approach will find many applications in labs-on-a-chip fabrication. Second, it is possible to use the self-assembly abilities of synthetic and biological molecules. Self-assembled nano- and microstructures can be generated from natural amphiphilic molecules, and these molecular self-assemblies may be used directly as potential biomimetic systems (for more details see ref. 38). This approach is more related to the fabrication of biochips to combine the sensitivity of a physical detection and the biological specificity. Because of the importance of DNA-based assays for detection of diseases, genome sequencing and environmental contol, a DNA chip is one of the major challenges.Supported layer
Self-assembled monolayers (SAMs) may be generated by the spontaneous physisorption or chemisorption of molecules onto a surface. Fusion of phospholipid vesicles on hydrophobic SAM, i.e., octadecyl trichlorosilane (Fig. 1), results in the formation of a phospholipid monolayer on a SAM (Fig. 2A). Peripheral enzyme membranes, such as pyruvate oxidase,39 and membrane electron carriers, such as plastoquinone and ubiquinone,40 can be incorporated in the supported bilayer at the physiological level (Fig. 1). When this bilayer was laterally in contact with a built-in gold electrode, lateral diffusion of quinone allows this electron carrier to shuttle between the electrode and pyruvate oxidase (Fig. 1). A catalytic current was then measured which reflected the electrochemical reaction of the quinone on the gold electrode, the lateral diffusion of the quinone along the bilayer and the catalytic reaction of the ubiquinone with the enzyme.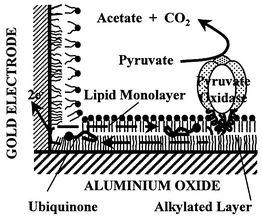 | ||
| Fig. 1 Reconstituted biological electron transfer. A lipid monolayer which incorporated a physiological amount of ubiquinone was added to the alkylated monolayer by fusion of lipid vesicles before the incorporation of pyruvate oxidase, a peripheral membrane enzyme. The electron carrier, ubiquinone, diffused along the biomimetic bilayer and made the shuttle between the gold electrode and pyruvate oxidase. The catalytic current measured both the electrochemical reaction of ubiquinone on the gold electrode, the lateral diffusion of ubiquinone along the alkane–lipid bilayer and the catalytic reaction of the ubiquinone with pyruvate oxidase. | ||
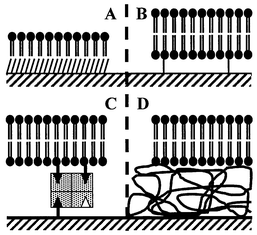 | ||
| Fig. 2 Schemes of supported lipid layer. A supported lipid monolayer that allows the incorporation of peripheral membrane proteins is obtained by fusion of lipid vesicles on the top of an alkylated layer (A). Several experimental methods were tested to obtain a lipid bilayer compatible with the incorporation of integral membrane proteins: lipid bilayer tethered by a hydrophilic spacer (B) or by an avidin–biotin complex (C); lipid bilayer onto a polymeric layer (D). | ||
During the last few years, technological efforts have been made to increase the efficiency of supported layers with comparison to the biological membrane. Formation of a new kind of lipid bilayer spaced from the surface by a linker has been done (Fig. 2). The bilayer was supported on a thin polymer layer41 (Fig. 2D) or tethered to the surface by a hydrophilic spacer42 (Fig. 2A), peptide43 or avidin–biotin complex (Fig. 2C). In the spaced bilayer, an integral membrane protein such as ATPase43 or rhodopsin42 has been successfully incorporated. Moreover, the spaced bilayer creates an ionic reservoir between the bilayer and the support.44 The ionic permeability of the bilayer can be modulated by the incorporation of peptides such as valinomycin44 and gramicidin45 that open the way to a new class of biocapture using ion-channel switches.46 We can guess that the future biochip based on a lipid bilayer will integrate membrane receptors to perform a very specific and very sensible analysis.
2D and 3D imprinting
For some purposes, nanostructures cannot be created by self-assembly alone. In these cases, a technique called ‘templating’ can be used in two possible ways: a template can be used to create a more complex initial pattern for subsequent self-assembly; alternatively, the original structure can be used as a template that can be modified by chemical or physical means to stabilize, or tailor, the properties for a specific purpose (for more details see ref. 38). Molecular imprinting is one example of the templating approach (Fig. 3). A ‘print’ molecule interacts with a solution of molecules (Fig. 3A), which is subsequently ‘fixed’ (Fig. 3B) to form an impression of the print molecule47 (Fig. 3C). Macroscopic polymer matrices are generally used to create the recognition sites (Fig. 3 upper part). The recognition sites of the polymer are related to a biomimetic approach to create synthetic receptors48 or synthetic antibodies.49,50 In some cases, the imprint has catalytic properties, such as ATP hydrolysis.51 Imprinting polymers appears then as a tool to create synthetic enzymes47 in order to increase the stability of the catalyst. The imprinting polymer matrices are applied mainly for affinity chromatography, i.e., for nucleotides,51,52 steroids,48 drugs49 or herbicide analysis.53 A thin film of the imprinting polymer can coat the surface of a physical transducer to create substrate-selective sensors. Examples of transducers used in imprinted polymer sensors are various: capacitance, amperometry, conductometry, potentiometry, pH, fluorescence, colorimetric, ellipsometry, surface plasmon resonance, surface acoustic wave and quartz microbalance.54 | ||
| Fig. 3 Schemes of 2D and 3D molecular imprinting. The different steps for 2D molecular imprinting (lower part) and 3D molecular imprinting (upper part) are: specific interactions with the host molecule (A), polymerization or self-assembly of molecules around the host molecule (B) and removing of the host molecule from the molecular imprint (C). The molecular imprint has both specific interaction sites and a specific shape to recognize the host molecule. | ||
Besides macromolecular polymers, 2D imprinting can be done using a SAM to create functionalyzed monolayers assembled on a surface, i.e., with size and shape specific molecular recognition sites (Fig. 3 lower part). Perforated monolayers that include molecular channels were then tailored and molecular components, i.e., tetradecanethiol and ‘trans’ quinone for photochemical imprinting,55 were co-assembled in two-dimensional monolayers.
DNA chips
DNA-based assays have an increasing importance for detection of diseases, genome sequencing, forensic and environmental control. They are challenged by the detection of trace amounts of DNA fragments, characterized by a specific sequence in a huge background of other molecules and unspecific DNA. The polymerase chain reaction (PCR) can be used in conjunction with labeled probes to detect very small amount of DNA. The following examples illustrate the purpose in a non-exhaustive way considering the huge research in the field of DNA chips.The diagnostic method is typically based on hybridization of oligonucleotides probes. Single DNA chips can consist of a matrix of dots coated with various oligonucleotides that simultaneously detect a range of DNA sequences. Deposition of DNA or oligonucleotides on the dot surface can be done by covalent linkage of activated oligonucleotide on amine functionalized surfaces,56 attachment of thiol functionalized DNA directly on a gold surface,57 electrochemical polymerization of oligonucleotide pyrrole,58 intercalation of DNA with a self-assembled monolayer of acridin,59 specific interaction of biotinylated oligonucleotide with streptavidin immobilized directly on the surface60 or on a lipid supported layer60,61 (Fig. 4).
 | ||
| Fig. 4 Schemes of oligonucleotide deposition. The deposition of oligonucleotide on the surface of a DNA chip can be performed in different ways: direct covalent linkage of the oligonucleotide on the surface (A), electrochemical polymerization of oligonucleotide pyrrole (C), immobilization with an avidin–biotin complex directly on the surface (B) or on a lipid supported bilayer (D). | ||
Sensors are mostly based on radioactive labeled probes, fluorescent probes or electrochemically active probes. Electrochemical probes can be obtained by the linking of an electroactive unit such as ferrocene62 with the oligonucleotide. The hybridization event can be also detected electrochemically by the intercalation of electroactive molecule such as Orange Acridin or daunomycin.63 A peak potential shift results from the hydrophobic interactions (intercalation) between DNA and intercalators. The properties of the intercalators to distinguish between double-stranded and single-stranded DNA are then very important to detect a gene specifically with high sensitivity. In the case of an oligonucleotide-functionalized polypyrolle electrode, hybridization decreases the intensity of the current peak due to the oxidation (or doping) of the polypyrrole chain.58 Using fluorescent probes, such as fluorescein labeled oligonucleotides,56 a simultaneous detection of multiple DNA sequences is possible using a fiber-optic biosensor array system. In this system, several optical fibers were bundled together each with a different probe immobilized on its tip. With the possibility to detect single fluorophore molecules, ultra-sensitive fluorescence techniques allow the detection of individual pairing of oligonucleotides.60
Another approach is based on the use of peptide nucleic acids (PNA)64 as a recognition layer in DNA chips. PNA is a structural DNA analogue with a pseudopeptide backbone that mimics DNA by forming complementary duplexes with normal DNA. The PNA/DNA duplexes, that can be done using shorter probes, have higher thermal stability and can be formed at low ionic strength.
C. Micro-total analysis systems
Micro-total analysis systems (μTAS), also called ‘labs-on-a-chip’, must perform the functions of large analytical devices in small units. They must contain elements for the acquisition, pretreatment, separation, post-treatment and detection of the samples.65 Biochips could be included as a functional element in a μTAS device but a number of other processes are necessary to go from reactants to analysis. Several methods exist for carrying out chemistry confined to well-defined regions of a planar device. For silicon or glass devices, methods include photolithography based on beams of light, electrons or ions; these methods have been typically developed for microelectronics. A network of microfluidic channels can be generated using an elastomeric polymer, e.g., poly(dimethylsiloxane),66,67 in which analytes are transported, mixed and separated in μTAS. Patterning of molecules on a reactive surface can be done by microcontact printing of the active molecule, e.g., alkanethiol on a gold surface,68 with an elastomeric stamp. In this case, patterning of the surface with a hydrophobic and a hydrophilic SAM can also create microfluidic channels. Conventional syringe pumps or microfabricated pumps perform liquid flow in the microfluidic microchannel. Release mechanism of drugs can be for example based on electrochemical dissolution of a thin anode membrane covering microreservoirs filled with chemicals in solid, liquid or gel form.69Strong points of the use of μTAS include the reduction of the consumption of samples and reagents, reduction of the time of analysis, and the increase of sensitivity. These high performances of μTAS will be particularly valuable in the pharmaceutical industry for the screening of combinatorial libraries, in clinical analyses, in DNA-based diagnostics and genotyping. Ramsey’s recent work using microchip electrophoresis as a separation method illustrates the performance. Separations of 15 pM Rhodamine 6G and 30 pM Rhodamine B (200 pL of solution) were performed using microchip electrophoresis and were detected by counting fluorescence bursts from individual molecules.70 A binary mixture of Rhodamine and dichlorofluorescein were resolved in less than 1 ms using microchip electrophoresis.71 Polymerase chain reactions were carried out on as many as four DNA samples at a time on a microchip device and the PCR products analyzed on the same device by microchip electrophoresis.72
D. Concluding remarks
The first major advances in nanotechnology were done by the microelectronics industry with the manufacture and machining of silicon materials. The second breakthrough was the emergence of the discipline of nanotribology from attempts to understand friction, wear and lubrication at the atomic scale, and the atomic-force and friction-force microscopes were developed as investigative tools for this purpose. The third step in nanotechnology is the convergence between the field and the recent achievements in biotechnology.There is not only a fertilisation of nanotechnologies through the use of biotechnologies but also a huge reciprocal impact of the nanoconcepts on biology.
The acquisition of expertise in these areas will soon become a prerequisite for those wishing to participate in the next phase of biotechnological evolution. Nanobiotechnology is important, not only for its research and development tools, but also for its potential commercial significance in the development of new analytical devices and preparative bioreactors.
References
- W. E. Moerner and M. Orrit, Science, 1999, 283, 1670 CrossRef CAS.
- S. Weiss, Science, 1999, 283, 1676 CrossRef CAS.
- J. K. Gimzewski and C. Joachim, Science, 1999, 283, 1683 CrossRef CAS.
- A. D. Mehta, M. Rief, J. A. Spudich, D. A. Smith and R. M. Simmons, Science, 1999, 283, 1689 CrossRef CAS.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930 CrossRef.
- T. P. Beebe, T. E. Wilson, D. F. Ogletree, J. E. Katz, R. Balhorn, M. B. Salmeron and W. J. Siekhaus, Science, 1989, 243, 370 CAS.
- N. B. Sheller, S. Petrash, M. D. Foster and V. V. Tsukruk, Langmuir, 1998, 14, 4535 CrossRef CAS.
- N. H. Thompson, B. L. Smith, N. Almqvist, L. Schmitt, M. Kashlev, E. T. Kool and P. K. Hansma, Biophys. J., 1999, 76, 1024 Search PubMed.
- D. Anafi and G. M. Wu, http://www.di.com/Theater/Misc/IgG-Amgen.html.
- A. P. Quist, A. A. Bergman, C. T. Reimann, S. O. Oscarsson and B. U. R. Sundqvist, Scanning Microsc., 1995, 9, 395 Search PubMed.
- M. Radmacher, M. Fritz, H. G. Hansma and P. K. Hansma, Science, 1994, 265, 1577 CAS.
- S. Kasas, N. H. Thompson, B. L. Smith, H. G. Hansma, X. Zhu, M. Guthold, C. Bustamante, E. T. Kool, M. Kashlev and P. K. Hansma, Biochemistry, 1997, 36, 461 CrossRef CAS.
- W. F. Heinz and J. H. Hoh, Trends Biotechnol., 1999, 17, 143 CrossRef CAS.
- S. P. Jarvis, H. Yamada, S. I. Yamamoto, H. Tokumoto and J. B. Pethica, Nature (London), 1996, 384, 247 CrossRef CAS.
- E. L. Florin, V. T. Moy and H. E. Gaub, Science, 1994, 364, 415.
- Y. S. Lo, N. D. Huefner, W. S. Chan, F. Stevens, J. M. Harris and T. P. Beebe, Langmuir, 1999, 15, 4373.
- S. S. Wong, E. Joselevich, A. T. Woodlley, C. L. Cheung and C. M. Lieber, Nature (London), 1998, 394, 52 CrossRef CAS.
- P. Hinterdorfer, W. Baumgartner, H. J. Gruber, K. Schilcher and H. Schindler, Proc. Natl. Acad. Sci. USA, 1996, 93, 3477 CrossRef CAS.
- S. Allen, X. Chen, J. Davies, M. C. Davies, A. C. Dawkes, J. C. Edwards, C. J. Roberts, J. Sefton, S. J. B. Tendler and P. M. Williams, Biochemistry, 1997, 36, 7457 CrossRef CAS.
- U. Dammer, M. Hegner, D. Anselmetti, P. Wagner, M. Dreier, W. Huber and H. J. Guntherodt, Biophys. J., 1996, 70, 2437 Search PubMed.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109 CrossRef CAS.
- D. W. Pohl, W. Denk and M. Lanz, Appl. Phys. Lett., 1984, 44, 651 CrossRef.
- A. Lewis, M. Isaacson, A. Harootunian and A. Murray, Ultramicroscopy, 1984, 13, 227 CrossRef.
- D. van Laeke, in Scanning probe microscopy; beyond the images, ed. S. Gauthier and S. Joachim, Les éditions de physique, Paris, 1992, pp. 229–275. Search PubMed.
- Ultramicroscopy, 1995, 61, pp. 1–314. Search PubMed.
- P. G. Haydon, S. P. Marchese Ragona, T. A. Basarsky, M. Szulczewxki and M. McClosky, J. Microsc., 1996, 182, 208 CrossRef CAS.
- T. Enderle, T. Ha, D. F. Ogletree, D. S. Chemla and C. Magowan, Proc. Natl. Acad. Sci. USA, 1997, 94, 520 CrossRef CAS.
- L. A. Gherber, J. Hwang and M. Edidin, Appl. Opt., 1998, 37, 3574 Search PubMed.
- M. F. Garcia Parajo, J. A. Veerman, A. G. T. Ruiter and N. F. van Hulst, Ultramicroscopy, 1998, 71, 311 CrossRef CAS.
- H. Muramatsu, N. Chiba, T. Umemoto, K. Homma, K. Nakajima and T. Ataka, Ultramicroscopy, 1995, 61, 265 CrossRef CAS.
- E. Betzig, P. L. Finn and J. S. Weiner, Appl. Phys. Lett., 1992, 60, 2484 CrossRef CAS.
- F. Zenhausen, Y. Martin and H. K. Wickramasinghe, Science, 1995, 269, 1083 CrossRef.
- M. Ishikawa, O. Yogi, J. Y. Ye, T. Yasuda and Y. Maruyama, Anal. Chem., 1998, 70, 5198 CrossRef CAS.
- H. P. Lu, L. Xun and X. S. Xie, Science, 1998, 282, 1877 CrossRef CAS.
- D. B. Craig, E. Arriaga, J. C. Y. Wong, H. Lu and N. J. Dovichi, Anal. Chem., 1998, 70, 39A CAS.
- H. Noji, Science, 1998, 282, 1844 CrossRef CAS.
- H. P. Erickson, Science, 1997, 276, 1090 CrossRef CAS.
- J. M. Laval, J. Chopineau and D. Thomas, Trends Biotechnol., 1995, 13, 474 CrossRef CAS.
- O. Pierrat, C. Bourdillon, J. Moiroux and J. M. Laval, Langmuir, 1998, 14, 1692 CrossRef CAS.
- D. Marchal, W. Boireau, J. M. Laval, J. Moiroux and C. Bourdillon, Biophys. J., 1998, 74, 1937 Search PubMed.
- J. Majewski, J. Y. Wong, C. K. Park, M. Seitz, J. N. Israelachvili and G. S. Smith, Biophys. J., 1998, 75, 2363 Search PubMed.
- S. Heyse, O. P. Ernst, Z. Dienes, K. P. Hofmann and H. Vogel, Biochemistry, 1998, 37, 507 CrossRef CAS.
- N. Bunjes, E. K. Schmidt, A. Jonczyk, F. Rippmann, D. Beyer, H. Ringsdorf, P. Graber, W. Knoll and R. Naumann, Langmuir, 1997, 13, 6188 CrossRef CAS.
- B. Raguse, V. Braach-Maksvytis, B. A. Cornell, L. G. King, P. D. J. Osman, R. J. Pace and L. Wieczorek, Langmuir, 1998, 14, 648 CrossRef CAS.
- A. T. A. Jenkins, R. J. Bushby, N. Boden, S. D. Evans, P. F. Knowles, Q. Liu, R. E. Miles and S. D. Ogier, Langmuir, 1998, 14, 4675 CrossRef CAS.
- B. A. Cornell, V. L. B. Braach-Maksvytis, L. G. King, P. D. J. Osman, B. Raguse, L. Wieczorek and R. J. Pace, Nature (London), 1997, 387, 580 CrossRef CAS.
- K. Mosbach, Trends Biotechnol., 1994, 19, 9 CAS.
- S. H. Cheong, S. McNiven, A. Rachkov, R. Levi, K. Yano and I. Karube, Macromolecules, 1997, 30, 1317 CrossRef CAS.
- R. J. Ansell, O. Ramstrom and K. Mosbach, Clin. Chem., 1996, 42, 1506 Search PubMed.
- M. Burow and N. Minoura, Biochem. Biophys. Res. Commun., 1996, 227, 419 CrossRef CAS.
- J. Mathew and O. Buchardt, Bioconjugate Chem., 1995, 6, 524 CrossRef CAS.
- K. J. Shea, D. A. Spivak and B. Sellergen, J. Am. Chem. Soc., 1993, 115, 3368 CrossRef CAS.
- J. Matsui, Y. Miyoshi, O. Dobhoff-Dier and T. Takeuchi, Anal. Chem., 1995, 67, 4404 CrossRef CAS.
- K. Haupt and K. Mosbach, Biochem. Soc. Trans., 1999, 27, 344 CAS.
- M. Lahav, E. Katz, A. Doron, F. Patolsky and I. Willner, J. Am. Chem. Soc., 1999, 121, 862 CrossRef CAS.
- J. A. Fergusson, T. C. Boles, C. P. Adams and D. R. Walt, Nature Biotechnol., 1996, 14, 1681 Search PubMed.
- R. Levicky, T. M. Herne, M. J. Tarlov and S. K. Satija, J. Am. Chem. Soc., 1998, 120, 9787 CrossRef CAS.
- H. Korri-Youssouffi, F. Garnier, P. Srivastava, P. Godillot and A. Yassar, J. Am. Chem. Soc., 1997, 119, 7388 CrossRef.
- N. Higashi, M. Takahashi and M. Niwa, Langmuir, 1999, 15, 111 CrossRef CAS.
- W. Trabesinger, G. J. Schutz, H. J. Gruber, H. Schindler and T. Schmidt, Anal. Chem., 1999, 71, 279 CrossRef CAS.
- K. Ijiro, H. Ringsdorf, E. Birch-Hirschfeld, S. Hoffmann, U. Schilken and M. Strube, Langmuir, 1998, 14, 2796 CrossRef CAS.
- S. Takenaka, Y. Uto, H. Kondo, T. Ihara and M. Takagi, Anal. Biochem., 1994, 218, 436 CrossRef CAS.
- K. Hashimoto, K. Ito and Y. Ishimori, Anal. Chim. Acta, 1994, 286, 219 CrossRef CAS.
- J. Wang, E. Palecek, P. E. Nielsen, G. Rivas, X. Cai, H. Shiraishi, N. Dontha, D. Luo and P. A. M. Farias, J. Am. Chem. Soc., 1996, 118, 7667 CrossRef CAS.
- B. H. Weigl and P. Yager, Science, 1999, 283, 346 CrossRef.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS.
- E. Delamarche, A. Bernard, H. Schmid, A. Bietsch, B. Michel and H. Biebuyck, J. Am. Chem. Soc., 1998, 120, 500 CrossRef CAS.
- E. Delamarche, H. Schmid, A. Bietsch, N. B. Larsen, H. Rothuizen, B. Michel and H. Biebuyck, J. Phys. Chem., 1998, 102, 3324 CrossRef CAS.
- J. T. Santini, M. J. Cima and R. Langer, Nature (London), 1999, 397, 335 CrossRef CAS.
- J. C. Fister, S. C. Jacobson, L. M. Davis and J. M. Ramsey, Anal. Chem., 1998, 70, 431 CrossRef CAS.
- S. C. Jacobson, C. T. Culbertson, J. E. Daler and J. M. Ramsey, Anal. Chem., 1998, 70, 3476 CrossRef CAS.
- L. C. Waters, S. C. Jacobson, N. Kroutchinina, J. Khandurina, R. S. Foote and J. M. Ramsey, Anal. Chem., 1998, 70, 5172 CrossRef CAS.
Footnote |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| This journal is © The Royal Society of Chemistry 2000 |