Analytical atomic spectrometry going into the next millennium: photons or ions, atoms or molecules?†
Alfredo Sanz-Medel
Department of Physical and Analytical Chemistry, Faculty of Chemistry, University of Oviedo, C/Julián Clavería 8, 33006, Oviedo, Spain
First published on UnassignedUnassigned7th January 2000
Abstract
Analytical atomic spectrometry seems to be suffering a sort of ‘identity crisis’ at the sunset of this millennium. The field has undergone profound changes and the rate of change has increased dramatically in the last decade. For many years the main goal of analytical atomic spectrometry was to provide atomic/elemental information on the composition of matter via the development of spectrochemical knowledge, instrumentation and determination strategies for the around 90 elements of the Periodic Table. Nowadays, however, we are witnessing a come back of many atomic spectroscopists to look for molecular (species) information, in a sort of re-encounter with ‘chemistry’ without renouncing the advantages of traditional spectrochemistry (sensitivity and selectivity). As expected, this turning point in the goals of the discipline seems to have been accompanied by profound changes in analytical tools and techniques used in this field. After reviewing the strengths and weaknesses of photon-based ‘workhorses’ of present routine elemental analysis, the dramatic change introduced by the combination of an ‘electrical’ flame (ICP) and a quadrupole mass analyser (MS) in a ‘recombinant’ instrument, the ICP-MS, is highlighted. The ICP-MS success originated a spectacular revival of atomic inorganic-mass spectrometry. Almost any possible coupling of a classical spectrochemical ion source with any mass analyser (developed for organic mass spectrometry) is now being intensively investigated. In fact, the popularity and economical importance of atomic MS has increased dramatically in the last few years and this importance is discussed in terms of techniques commercially available and those under active development. In order to cope with present needs for chemical speciation information, atomic techniques are widening their scope to be able to provide molecular information. One approach consists of studying new MS instrumentation capable of producing both atomic (elemental information) and molecular (molecular ions and fragmentation) mass spectra in the same instrument. Additionally, an extra degree of specificity can be afforded by coupling powerful separation techniques (gas or liquid chromatography, capillary electrophoresis, etc.) to the above spectrometric instruments, which would eventually be able to provide elemental, molecular and perhaps structural information of the compound(s) responsible of a given chromatographic or electrophoretic peak. In brief, more ambitious goals, more powerful elemental detectors (based on both photon and ion measurements), more and more flexible hybrid techniques and more active cross-fertilization with other fields of science are good indicators of a bright future for analytical atomic spectrometry entering the next millennium.
1 Evolution and natural selection of analytical techniques
Just as in biological evolution, a scientific topic which I enjoy as an ‘amateur’, environmental pressures and survival needs trigger recombination mechanisms and final ‘mutations’ of analytical techniques, which seem to be continuously re-adapting/renewing in order to accommodate to new environment conditions.Environmental pressures might come from scientific curiosity or, more and more these days, from chemical information needs posed by modern science, technology or compliance with social regulations. Survival needs originate a sort of ‘natural selection’ of the toughest techniques (in contrast to the plethora of techniques developed and published in the analytical literature) and, in the long run, ‘mutations’ can occur which, more or less reluctantly, could be eventually accepted and installed in the routine analytical laboratory.
Environmental pressures can be both purely scientific and application-oriented unsolved problems. They are the real driving forces of analytical research (at the basis of analytical evolution) which paves the way to either predictable ‘recombination’ mechanisms or not so predictable ‘mutations’ occurring in the history of analytical instrumentation. Analytical research is continuously developing. A given analytical technique, however, is established only after a rather peculiar process: first, a physical or chemical principle is shown to be useful for chemical analysis; the phenomenon is thoroughly investigated, characterized and understood; then, an adequate instrument to measure the corresponding ‘analytical signal’ is developed and improved; finally, applications and methodologies are widely developed. Of course, new techniques could emerge (as biological new species appear) when a previously unexploited principle is demonstrated as being successful for chemical analysis, but this ‘mutation’ is unpredictable. Recombination or hybridisations of existing techniques are more likely and predictable (sooner or later someone will do it). As pointed out by Dawson1 ‘timeliness’ is a key factor for the final success of a technique; the real need creates the driving force for research and the basic knowledge and the technological development should be coincidentally there. I should also add the timeliness of the entrepreneurial initiative of a company convinced that commercialization of the technique/instrument/applications can be good business. For a company to embark on such economic venture the following points should be verified:2 the market exists, the technique will be accepted by potential users, there is a know-how for production and commercialization of the instrument and, most importantly, the company will make a profit. Timeliness is thus decisive again. The final proof for success of a given analytical ‘recombination’ or ‘mutation’ is commercialization which would promote eventually worldwide acceptance. For instance, ICP-MS was a most powerful analytical tool invented back in 19803 but its worldwide exponential growth we are witnessing nowadays4 is only possible because of its commercialization in 1983 and progressive acceptance during that decade.
More than ever before, natural selection (success) of a given analytical ‘recombination’ or ‘mutation’ seems to be tied to, and assessed by, economical indicators. Only a timely commercialized instrument of great analytical potential seems to have opportunities to become a real routine ‘workhorse’ for analysis and, in doing so, it gives back the expected profits out of the original investment. In contrast, formerly well established techniques can become ‘extinct species’ if economical timeliness is passed over.
Thus, in attempting to assess the importance of analytical atomic spectrometry in the frame of modern analytical chemistry5 perhaps its relative economic importance could provide a rather significant indicator.
The worldwide market of instrumentation has been estimated to amount to around 13,200 millions US dollars for 1998 and it appears that elemental analysis by atomic techniques would amount to 1,100 millions. This represents 9% of the total market worldwide.2 Of course, there are real ‘workhorses’ for routine elemental analysis (see next section) accounting for most of the 1,100 millions US dollars sales, still a very attractive business. This relative success derives from the fact that atomic spectrometry based techniques have been continuously evolving in order to be able to dominate (as they do nowadays) the elemental inorganic analysis. It appears that via ‘mutations’ (e.g., the use of plasmas introduced in the seventies) and ‘recombinations’ (e.g., ICP-MS) the survival of atomic techniques in analytical instrumentation seems secured. Fig. 1 shows current and projected market size, in millions of US dollars, for ICP-OES and ICP-MS instrumentation, according to Montaser.4
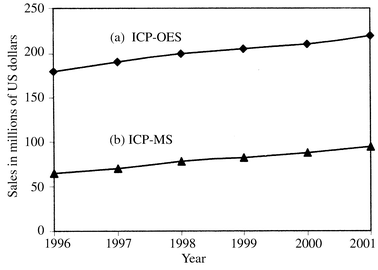 | ||
| Fig. 1 Current and expected worldwide market size for ICP sales: (a) ICP-OES, (b) ICP-MS instrumentation (adapted from Fig. 6 of Ref. 4). | ||
Analytical evolution seems to proceed as in bacterial populations, that is, maintaining part of the acquired genetic integrity.6 This is acquired basic analytical knowledge and technologies. Both should be available to facilitate recombinations and perhaps to promote eventual mutations of those established techniques which fail ‘to accommodate’ to new environment demands.
Thus, it seems advisable to review first the strengths and weaknesses of the well established atomic spectrometric techniques which evolved to become ‘workhorses’ for elemental routine determinations (photons-based techniques). Then, we will review the increasing importance of emerging techniques based on ion measurements.
However, from the general perspective of analytical chemistry, atomic methods only represent about 10% of total analytical determinations (a figure coincident with relative figures in a modern clinical laboratory7). In other words, atomic information is useful, but molecular information is more and more demanded. It seems that evolutionary changes in the field of analytical atomic spectrometry are particularly evident in this respect. Atomic detectors are now ripe to contribute effectively to molecular analysis as well. In fact, we are witnessing a tremendous research effort to expand the traditional elemental scope of atomic methods into the realm of molecular information as well.8
Going into the next millennium happens to be a time for fast evolution. Therefore, perhaps my attempt to put present analytical atomic spectrometry into place can be a positive exercise for the readers, in spite of the unavoidable personal perspective of such an undertaking.
2 Current workhorses for inorganic elemental analysis
Routine inorganic elemental analysis is mainly carried out nowadays by atomic spectrometric techniques based on measurements of ‘photons’ (e.g., based on interactions between photons of electromagnetic radiation and the matter to be analyzed). Current workhorses for such analysis include both, dissolved sample and direct solid analysis techniques.Dissolved sample analysis techniques
Flame-AAS (FAAS), electrothermal atomization (ET-AAS) and inductively coupled plasma-OES (ICP-OES) are probably today’s ‘workhorses’ for dissolved sample routine analysis.Instrumental development as well as analytical applications, which increased after extensive commercialization of AAS in the sixties and of ICP-OES in the eighties, have been profound and extensive along the years. Knowledge about such techniques is so well advanced by now that it seems reasonable to assume that new spectacular breakthroughs are not likely. Capabilities and analytical limitations are well known in comparing their respective analytical performance characteristics. An attempt in that direction is summarized in Table 1 which gives a general comparative assessment, in the light of many years of development, of the main ‘pros’ and ‘cons’ of each of these three popular techniques.
| Flame-AAS | Electrothermal-AAS | Inductively coupled plasma-OES | |
|---|---|---|---|
| Advantages | The most widespread and known. | Very low DLs (102–103 better). | Multi-elemental (simultaneous). |
| Simple, inexpensive and reliable. | Adequate for ultratrace (μg L−1). | High atomization temperature. | |
| Moderate (and well known) interferences. | Microsample analysis. | No refractory compounds problems. | |
| A ‘problem solver’ for small labs. | Specially suited for life sciences analysis | Very low matrix-interferences. | |
| Limitations | Uni-elemental. | Uni-elemental. | Serious spectral interferences. |
| Moderate DLs (in the mg L−1 range). | Discontinuous operation. | Comparatively moderate DLs (except for axial configuration). | |
| Low atomization temperature (refractory compounds problem). | Carbide formation and comparatively slow. | More expensive to run. | |
| Limited for non-metal analysis. | Limited for non-metal analysis. | Rather limited for non-metals. |
Taking the risk inherent in excessive generalizations, perhaps we could say that AAS dominates elemental inorganic analysis carried out in rather small laboratories and when only a few analytes (probably at mg L−1, ppm, levels) have to be determined.
When ppb level (μg L−1) sensitivity is required the technique of choice is ETAAS, at a cost of simplicity, robustness and speediness of the analysis as compared to FAAS.
ICP-OES appears to have become the most popular routine technique for inorganic multielemental analysis of dissolved samples,9 even if initial investment and subsequent running expenses are much higher than those needed for AAS. The 1998 worldwide market size for ICP-OES amounted to around 200 millions US dollars and a steady increase of that market for the next three years is foreseen.4 In contrast, by this time direct current plasma (DCP)-OES has become ‘extinct’. Microwave induced plasma (MIP)-OES, however, has evolved to become a successful specific detector for metals, semimetals and non-metals, although samples should be presented as a vapour or gas to the atomizer, as it is case in gas chromatography–MIP-OES.10 Of course, flow injection analysis (FIA) strategies have become commonplace in most laboratories opening new perspectives to sample handling, pretreatments and manipulation for atomic spectrometry methods.
Direct solid analysis techniques
The rapid growth of ICP-OES might obscure the fact that the market size for direct solid analysis is still equivalent to that of the dissolved samples as Mermet2 pointed out recently (Arc/Spark-OES still represents a worldwide market of 210 millions US dollars similar to that of ICP-OES).Spark source-optical emission spectrometry (SS-OES) and X-ray fluorescence spectrometry (XRF) are very well established routine techniques which play a most important role in industrial control analytical chemistry of solid materials. Their importance today to monitor industrial processes, metallic and non-metallic raw materials and final products, cannot be overemphasized. They have no competitors in that application. Both techniques are so well implemented in routine laboratories that it is sometimes said that there is nothing new in X-ray or spark source based analysis.
However, modern SS techniques allow broader areas of the sample electrode to be vaporized, lower detection limits to be achieved with great accuracy and precision11 and they are normal accessories today for solid sample introduction into plasmas. X-ray analysis cannot be considered obsolete either: apart from novel X-ray methods based on synchrotron radiation sources,12 there is an important activity on quantitative analysis using the electron microprobe13 and also a distinct interest to develop ‘portable XRF’ instruments for the direct in situ analysis of the samples.14
Of course, total (TXRF reflection X-ray fluorescence) continues to expand while its applications to environmental samples and surface analysis (e.g., Si wafers) are growing steadily.15 However, TXRF cannot be considered today an analytical ‘workhorse’, nor direct-solid analysis its major application.
Conversely, laser induced breakdown spectroscopy (LIBS), laser ablation with plasma sources (LA-PS), sparks with plasma sources, electrothermal vaporization of slurried samples (for ETAAS, but also for plasma sources) are now being developed actively and could become future workhorses for direct solid analysis (as accessories for plasma sources).
3 Alternative detection systems: mass analyzers versus photon detectors
The classical instrument of atomic spectrometry (Fig. 2) has been built using the interaction, in the spectrochemical source, of the atoms to be analyzed with UV-VIS photons which were to be measured. Absorption, emission or fluorescence ‘spectra’ obtained provided the qualitative and quantitative information needed for the elemental analysis. Photon detection thus is crucial and, with the years, the photomultiplier became the most popular UV-VIS transducer.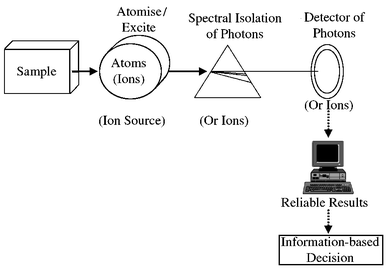 | ||
| Fig. 2 A general schematic of an ‘atomic spectrometer’. | ||
Research in spectroscopic detectors continued to fuel evolution and allowed to improve measurements in this region of the electromagnetic spectrum. In fact, detection of plasma (usually an ICP) emissions by modern charge transfer devices (CTDs) has been most successful during the last five years or so.16–18 When these ‘array detectors’ are coupled with échelle spectrometers a most powerful detection system results for UV-VIS emissions from plasmas: now the favorable features of the old photographic plate (particularly simultaneous detection of multichannel signals and background) are accessible with modern electronic signal transduction and processing. As pointed out by Denton et al.16 most of the desirable properties of an ‘ideal’ atomic optical emission detector can be approached rather closely by CTD-OES available systems, run in the scientific mode (see Table 2). Successful commercial instrumentation for ICP-OES measurements has been developed using both, ‘charge injection devices’ (CIDs)16 and ‘segmented charge coupled devices’ (SCDs).17,18 Both multichannel array detectors provide an extremely rich spectral information which is becoming more and more popular for routine elemental analysis by ICP-OES.
| Desirable properties of OES detectors | MS detectors properties |
|---|---|
| High sensitivity (μg L−1) | Very high sensitivity (ng L−1) |
| High resolution power (λ/Δλ) | Capabilities of up to 10000 mass resolution power (m/Δm) |
| Wide wavelength coverage | Elemental mass coverage 1–250 u |
| Low detector noise | Low detector noise |
| Absence of dark current | Low background (off peak) |
| Linear response and wide dynamic range (4–5 orders) | Wide linear range (6-8 orders) |
| Multichannel detection | Possibility of truly simultaneous multielemental determinations (e.g., multicollectors) |
| Simultaneous background correction | Not needed |
Of course, other developments in ICP-OES are under scrutiny, apart from exploitation of multichannel detectors,16 and I would stress: advantages of axial rather than lateral viewing of the plasma,19 development of convenient softwares for fast semiquantitative analysis,20 boosting of basic studies to describe adequately ICP excitation mechanisms and diagnostic tools,21 advanced sample introduction systems into plasmas (e.g., lasers,22 sparks,11 volatile species generation23), the use of organized media to improve atomic spectroscopic methods24 and others. However, ICP-OES became a mature technique a decade ago9 and is now the benchmark tool for inorganic multielemental analysis.
AAS techniques are very well established by now. This along with ruthless competition of plasma spectrometry makes novel research and development comparatively scarce. Of course, there are reports on new applications of ETAAS to ultratrace analysis and some new developments in atomisers, but future atomiser design developments will probably involve only minor modifications.25 Perhaps more exciting attempts will be to use diode-laser based AAS with different modulation improvements and exotic atomizers able to analyze non-metals with excellent detectability.26,27 In the instrument exhibition of ‘Analytica, 98’ (Germany) a commercial instrument based on using such diode-lasers instead of HCLs has been already introduced (mainly for semiconductors elemental analysis). Moreover, it has been claimed very recently28 that the future of AAS lies ahead on simultaneous multielemental analysis carried out with continuous source lamps in connection with modern multichannel array detectors. Of course, ICP-OES and ICP-MS potential for simultaneous multielement analysis seems to be a great handicap to allow widespread commercial use of such advanced AAS systems. It is more likely that modern solid sampling ETAAS,29 if heterogeneity in the samples for the sought elements is taken into account, could provide practical alternative ‘niches’ to new AAS applications.
Mass analyzers versus photon detectors
Evolution via heterogeneous recombinations is a known mechanism in biology6,30 which seems to have a ‘parallel’ in analytical atomic spectrometry. Alkemade31 predicted in 1973 that ‘flame ionic mass spectrometry’ could be a favorable extension of the already developed flame atomic absorption/emission methods. Orders of magnitude improvement in detection limits could be expected changing the detection mode from one for photons to one for ions of interest. The practical realization of this idea came in a different form, however, when an ‘electrical flame’, the Ar-ICP (very well developed and reknowned as an exceptional spectrochemical excitation source for ICP-OES), was used for the purpose. It was in 1980 when Fassel’s group demonstrated for the first time the successful interfacing of an Ar-ICP with a quadrupole mass spectrometer.3 Basic and applied developments of this ‘recombinant’ ICP-MS technique took place at such a rate during these years4 that a total number of around 2,500 ICP-MS instruments are estimated to have been sold by now2 in spite of the discouraging price of such instruments ranging from $140,000–180,000 for a conventional quadrupole analyzer to $400,000–800,000 for high resolution double focusing and multicollector instruments.4 Timeliness must have played a crucial role for the success of such a ‘recombination’ because both, ICP and quadrupole-MS technologies, were well advanced by the time. Instrument development seems to mimic nature’s evolutionary mechanisms (recombination, mutation and natural selection) to find and build instruments for specific tasks with occasional revolutions within long-term evolution.30 In fact, ICP-MS instruments, ‘a sort of revolution in natural evolution of instrumentation for elemental analysis’, are even closer to the ‘ideal’ instrument for multielemental analysis32 than ICP-OES. ICP-MS analytical performance characteristics today are so striking (see the comparisons of Table 2 with ICP-OES and in more general terms in Table 3) that one could wonder if this technique, borne as ‘another application’ of ICP, will not bring about eventually commercial extinction of its ancestor, ICP-OES (of course, that extinction will not happen in the very near future according to Fig. 1).| ICP-MS and ICP-OES |
|---|
| Multielemental character (simultaneous multielement analysis possible). |
| Speed of analysis. |
| Semiquantitative rapid analysis (easier by ICP-MS). |
| Continuous operation (on-line detector for chromatography and FIA). |
| ICP-MS versus ET-AAS |
| ET-AAS does not exhibit any of the above features. |
| ICP-MS provides today better sensitivities (102–103 times superior). |
| ICP-MS exclusive features |
| Specific elemental detection in the ng L−1 range (or below). |
| Isotope measurement capability allowing for: |
| Reliable confirmation, by several isotopes, of the presence of a given element. |
| Isotope dilution (‘definitive’ methods development). |
| Stable isotope tracers applications (e.g., to follow the metabolism of elements). |
| ‘Speciated’ ID-MS methods for validating speciation results. |
Probably the most differential feature of ICP-MS versus photon-based measurement techniques is ICP-MS ability to determine isotopes and to measure readily isotopic ratios. This capability gives to ICP-MS exclusive applications including: (a) the flourishing of isotope dilution (ID)-ICP-MS methods for accurate determinations33 of trace and ultratrace levels in the most varied samples;34 (b) the development of ‘speciated’ ID-ICP-MS methods in order to validate results of species or compounds of a given trace element obtained by using hybrid techniques (e.g., in environmental35 and in biological materials;36 (c) the flourishing of ICP-MS methods to measure ‘stable isotopes tracers’, administered to living organisms in order to follow the metabolism of essential and toxic elements. Traditionally, radioactive tracers were employed for this purpose and most of the present knowledge about metal metabolism derives from experiments based on the administration of the corresponding radioactive isotopes to the studied living organism. Their eventual transport and distribution in the different organs is followed by radiochemical measurements. Of course, application of this radiochemical technique in humans has always been hindered by the detrimental effects of γ radiation to the body. With the advent of ICP-MS a parallel strategy using non-radioactive isotopes seems straightforward.37,38
To conclude this section, however, it must be pointed out that ICP-MS is still a comparatively expensive analytical tool with a high price and maintenance costs. Moreover, its adequate running demands highly qualified personnel so far. Both factors hinder its widespread use in many routine laboratories. However, its striking analytical performance characteristics (see Table 3) allows us to foresee a gradual introduction of ICP-MS instruments not only in research but also in routine analytical laboratories from the year 2000 and onwards (see Fig. 1 for a glimpse of the growing of market size for ICP-OES and ICP-MS up to the year 2001).
4 The revival of atomic (inorganic) mass spectrometry (At-MS)
In contrast to the great success of organic mass spectrometry, inorganic (atomic, elemental) mass spectrometry has been comparatively stagnant during the last fifty years, considering that the first analytical instrument, a spark source-MS one, was introduced in the fifties.39 The unprecedented success of the ‘recombinant’ ICP-MS technique (and to a lesser extent of glow discharge (GD)-MS) has brought about the revival of atomic mass spectrometry (At-MS) in atomic spectrometry.Within the ICP-MS field we are witnessing already the third generation of quadrupole-based instruments and seeing also active developments of alternative mass analysers. At the same time, glow discharges (GD) continue to attract much interest for the direct solid analysis by GD-MS. Moreover, fields of GDs applications for non-conducting40 or concentration profile analysis of layered materials41 or the use of pulsed-GDs coupled to TOF mass analysers for improved analytical performances42 are being developed and exploited. Many of the techniques developed in this half of the century for At-MS (see Table 4 to get a glimpse of the great variety of such techniques) seem to have been born again following the present success of ICP-MS.
| Source | Sample | Technique (name) | Acronym |
|---|---|---|---|
| a Techniques which can be used for analysis of some special inorganic compounds, e.g., organometallics, though no atomic ions formation is at the basis of the analysis. | |||
| Filament | Liquid | Thermal ionization mass spectrometry | TIMS |
| ICP | Liquid | Inductively coupled plasma mass spectrometry | ICP-MS |
| Glow discharge | Solid | Glow discharge mass spectrometry | GD-MS |
| Spark source | Solid | Spark source mass spectrometry | SS-MS |
| Laser | Solid | Laser ionisation mass spectrometry | LIMS |
| Ion beam | Solid | Secondary ionisation mass spectrometry | SIMS |
| Ion beam | Solid | Sputtered neutral mass spectrometry | SNMS |
| Ion beam | Solid | Accelerator mass spectrometry | AMS |
| ETA-MIP | Liquid | Furnace atomisation mass spectrometry | FAPI-MS |
| Electrospraya | Liquid | Electrospray mass spectrometry | ESMS |
| Fast atom bombardmenta | Liquid | Fast atom bombardment mass spectrometry | FABMS |
In atomic mass spectrometry the mass spectrometric measurement of atomic ions (as opposed to ‘molecular’ ions or fragments used for organic MS) is usually performed in order to determine the elemental or/and isotopic composition of the sample. Thus, all the At-MS techniques rely on the use of a mass spectrometer able to analyse a beam of gaseous ions with different masses and separate them according to their mass to charge ratio. That is, the spectrometer plays a role similar to that of the monochromator in conventional photon spectrometry, except that masses rather than photons (frequencies) are separated and measured. In this way, however, much higher sensitivities for trace element determinations and also isotopic composition capabilities are at hand.
Mass spectrometers used in At-MS are similar to those developed for organic mass spectrometry. Only the required mass range (from 1 to 250 in usual atomic applications) and abundance sensitivity will basically change. Of course, the quadrupole mass filter is the most popular MS analyser due to its relatively low cost and easy handling. However, the main problem of the quadrupole is its limited resolution in the mass spectrum (about 1 u, as commonly used). Although the number of lines are much less in the mass than in the optical spectra, spectral interferences are still very serious in At-MS work.
These interferences, resulting from the plasma gases, from interelemental isobaric and polyatomic spectral overlaps or from matrix-induced interferences, are very serious and are creating a great deal of research activity: previous separations, aerosol pretreatments, mixed plasma gases, ‘cool’ plasmas, gas-phase reaction chemistry in de-clustering collision and reaction cells, etc., are different approaches aiming at eliminating or reducing such interferences.43 The more common approach to date is to resort to high resolution instruments able to separate isobaric interferences from polyatomic ions. The most popular high resolution instruments today use an electrostatic analyser in combination with a magnetic sector.44 This combination provides double focusing, that is angular (m/z) and kinetic energy (1/2 mv2) focusing. Ion deflections by small energy differences caused by the magnetic sector are corrected by an exactly inverse deflection by the electrostatic analyser. Thus, ions with a given m/z value and different kinetic energies will be finally deflected to the same point by the double focusing (DF, which is obtained for a given mass at a given moment) and the instrument should be named ICP-DF-MS.
Resolving power (M/ΔM) values up to 10000 can be obtained in such instruments, able to separate adequately many of the spectral overlaps from polyatomic ions. At low resolution the sensitivity attainable can be 10–100 times better than in quadrupoles. Thus, applications of ICP-DF-MS are rapidly growing in the areas of ultratrace total element determinations, precise isotope ratio measurements analysis and trace metal speciation.45
Of course, operation of quadrupoles in higher regions of stability has been demonstrated to offer also higher resolution for ICP-MS than conventional operation, although significant compromise in sensitivity is required (as it is the case for sector instruments as well).46 Apart from quadrupoles and double focusing MS instruments, time of flight (TOF) MS analysers seem to be roaring in the last two years, after the commercialization of both orthogonal47 and axial48 ICP-TOF configurations, introduced by two different companies and most actively investigated by Hieftje et al.47,48
Other mass analysers, less common in At-MS, are under investigation as well. So Koppenaal et al.49 are pursuing the development of plasma source-ion trap (PSIT). In this development of the quadrupole, transiently formed ions (e.g., after laser ablation) could be stored before their final ejection from the trap to the detector.50 Gas phase ion–molecule reaction methods to remove isobaric interferences in PSIT, not through higher mass resolution but via highly selective gas phase reactions,49 have been reported. For extreme mass resolution, the development of Fourier transform ion cyclotron resonance (FT-ICR) mass analysers should be mentioned here.51 The highest previously reported mass resolving power in ICP-MS was 46000 at ppm levels of the analyte for a magnetic sector based instrument, while the recently developed ICP-FT-ICR-MS developed in Florida University seems to be able to provide baseline separation of Ca+ from Ar+, with a calculated resolving power of 280 00052 at ppb levels.
Ion detection (photographic in the early days of SS-MS), is carried out electronically with a secondary electron multiplier (SEM) or a Faraday cup. Channeltrons have also been popular but they have shorter lifetimes, due to total accumulated charge, damaging the PbO semiconductor. Nowadays, there is a tendency to replace continuous dynode channeltrons by the discrete SEM devices. The Faraday cup is a very simple transducer, basically made of a hollow metallic conductor. It is scarcely affected by mass discrimination effects or by different energies of the separated ions. Thus, the Faraday cup can be a favorable ion transducer for measuring high MS signals with great precision.
The typical single-channel scanning operation of a magnetic sector, with a 50–100 μm fixed slit, can be converted (as in polychromators) to simultaneous multichannel detection by removing the collector slit and: (a) adding several slits and transducers in strategic positions for simultaneous ion collection53 or (b) adding a multichannel detector as the diode array or a CCD to measure the separated ions. While the multicollector approach has been already developed commercially,53,54 the second one is under investigation.16 Of course, such multichannel arrays could provide high-precision measurements of isotope ratios. The advantages of speed and easy sample handling provided by the ICP (e.g., versus TIMS measurements) could be achieved for high-precision isotope ratio measurements applications by incorporating multicollector MS to conventional ICP ion sources.53
In principle, any combination between every spectrochemical/ion source developed so far and any known mass analyser, such as those mentioned above, could be possible (see Fig. 3). In fact, many of the possible combinations from Fig. 3 have already or are being investigated. Tandem sources as furnace atomisation plasma ionization (FAPI), are also now being explored for MS detection.55 In any case, in practice, only a few of such combinations have become commercial instruments. Table 5 summarizes the more common combinations along with their main analytical features and their most relevant application. The last technique in Table 5 (laser based sample introduction techniques coupled to a MS-TOF detector) deserves special mention at this evolutive moment. This particular arrangement (Fig. 4) has been extremely successful for biomolecules analysis (e.g., MALDI-TOF techniques) and could be one of the analytical ‘workhorses’ of the near future for elemental direct solid analysis, using an ICP-MS (TOF) detector.
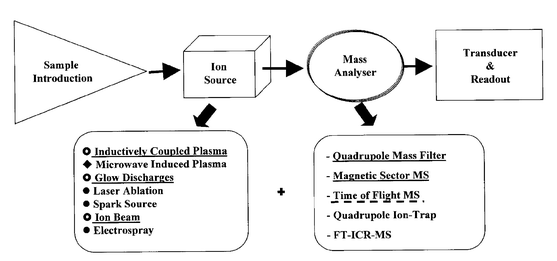 | ||
| Fig. 3 In the search for the ‘ideal’ instrument for atomic mass spectrometry (At-MS). | ||
| Technique | Main features and field of application | Recommended references |
|---|---|---|
| TIMS | Reference technique for precise isotope ratio measurements. | 39 |
| Comparatively laborious and slow. | ||
| Dissolved samples. | ||
| Excellent for sensitive, rapid, multielement analysis. | ||
| ICP-MS | Poorer precision for isotope ratio measurements. | 32, 56, 57 |
| Basically a technique for dissolved- sample analysis. | ||
| Alternative to ICP-MS for direct solid analysis. | ||
| GD-MS | Good detection limits in the solid (μg–ng g−1). | 39, 40 |
| Conducting and isolating materials analysed. | ||
| Surface and interphase analysis (qualitative). | ||
| SIMS | Very good sensitivity in the solid (ng g−1). | 39, 58 |
| Exceptional lateral resolution (1–0.1 μm). | ||
| Localised microanalysis (qualitative). | ||
| LIMS | High detection power (about 10−20 g). | 39, 59 |
| Microbeam character (spatial resolution about 1 μm). |
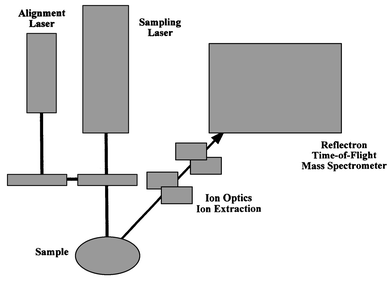 | ||
| Fig. 4 Schematic of laser-assisted sample introduction with final TOF-MS detection of generated ions in the laser pulse. | ||
5 Trace element speciation: hybrid techniques
Total trace element determinations of metals and semimetals in virtually any type of material are increasingly demanded. Such determinations of very low levels of the elements are normally accomplished by atomic spectrometric methods because of their sensitivity and specificity. However, in the last two decades, the scientific community has recognized that toxicity, bioavailability, bioactivity, metabolism, transport, persistence, final fate, bio-geological cycles and, thus, the eventual impact of a toxic element in our body and environment will be dictated by its particular form (species) present in the sample. In this sense total element determinations by atomic spectrometry, as discussed before, are not sufficient today to assess trace element roles (moreover, total contents might be misleading, e.g., AsIII is toxic but arsenobetaine is not). In brief, additional ‘speciation’ information to complement total elemental determinations is being increasingly demanded in environmental science, biology, medicine, eco- and clinical toxicology, occupational health and hygiene, food and energy industries.6,60,61The present importance of analytical speciation information can be judged by the fact that about 300 papers per annum have appeared in the last two years62 and well over half a dozen books have been published so far dealing with speciation issues.63–66 Moreover, legal regulations agencies seem to acknowledge and recognize progressively speciation information importance.67 Speciation information is demanded for many chemical species and elements in the most varied fields (see Scheme 1 for illustration). Thus, there is an enormous interest now to develop analytical strategies and methods able to tackle this modern challenge of ‘speciating’ trace element concentrations.
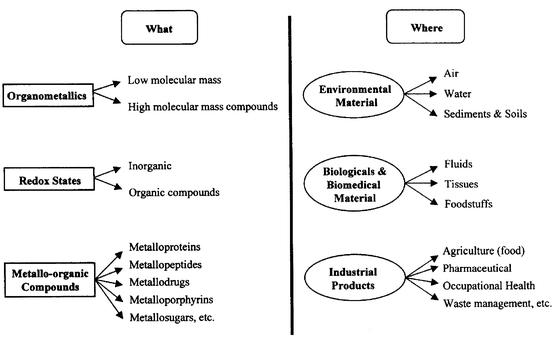 | ||
| Scheme 1 What and where to speciate? | ||
Of course, atomic methods are by definition non-speciating methods because the atomiser destroys the molecules and so any molecular (species) information. Thus, new instrumentation, analytical approaches and methods are needed able to provide also reliable molecular information of the species of a given trace element in a real sample. Whereas an array of reliable instruments for routine total trace element determinations is readily available in the market, instrument manufacturers are still reluctant to support speciation analysis.62 In fact, analytical speciation is carried out today almost exclusively in research laboratories, although a progressive extension to routine laboratories is clearly envisaged.
Four different approaches or general tools to face the modern challenges of trace element speciation can be identified:61 (a) computational approach; (b) direct species-specific techniques (e.g., bio-sensors); (c) hybrid or hyphenated techniques; (d) physicochemical characterization techniques as employed for bioinorganic chemistry.68
Among them, however, it is undebatable that hybrid techniques (that is the coupling of a powerful separation unit with an element-specific atomic detector) are preferred to solve real-life speciation problems. That is, the coupling of a separation technique (GC, HPLC or even capillary electrophoresis) with an adequate atomic detector, ideally operating in a continuous manner for on-line real-time element-specific detection, seems to be at present the most versatile, useful and popular approach. The selection of the separation technique will depend on the nature and physical properties of the species to be determined in a given sample. The two more general hybrid approaches used, along with the main criteria to select the analytical speciation strategy depending upon the features of the trace element species to be determined, are shown schematically in Scheme 2.
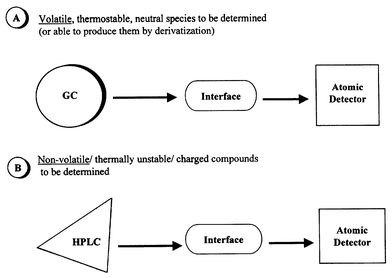 | ||
| Scheme 2 The two most common hybrid approaches for the speciation of a given form of the trace element under study. | ||
Traditionally organic mass spectrometry has been aware of the need to separate compounds before final identification and detection. Hybrid techniques such as GC-MS or LC-MS are widespread for identification, confirmation and/or determinations of species (compounds) derived from non-metals (C, H, N, O). Such molecular and/or structural information is now needed for the speciation of toxic and essential metals and semimetals forming part of organometallics, biomolecules, etc. In other words, the traditional distinction between atomic and molecular analysis is becoming blurred for analytical spectroscopists engaged in speciation.
As these traditional conceptual boundaries blur, the corresponding techniques to get modern trace element information are quickly evolving too. This can be observed in present trends in MS instrumentation being developed for speciation: present ion sources for MS essentially fall in two categories, namely ‘organic’ ones, creating molecular or fragment ions (e.g., electron impact, chemical ionization or fast atom bombardment), and ‘atomic’ ones which produce atomic ions (ICP, MIP or GD). However, in the last years different MS systems under development appear to be capable of both element specific detection and fragmentation to molecular ions simply by adjusting the discharge parameters of the ion source. Low pressure ICP-MS,69 electrospray ionization TOF-MS70 and GD-MS71 have been proposed, although further research and development appear to be needed.
Electrospray (ES) ionization is a technique widely used in organic analysis with the aim of obtaining ions directly from a solution (see Fig. 5) and has become a successful HPLC–MS interface.72 ESMS (Table 4) is becoming an enlightening example of this blurring of inorganic/organic (atomic/molecular) boundaries. ESMS, as pointed out in Table 1, is a ‘soft’ ionization technique, but it is being investigated for inorganic mass spectrometry showing a great promise for speciation studies.73
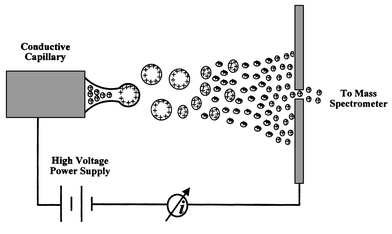 | ||
| Fig. 5 Schematic diagram of the electrospray ion formation process in ESMS. | ||
Additionally, and considering the complexity of real-life speciation problems, an extra degree of specificity can be afforded again by coupling powerful separation techniques to the above spectrometric instruments which would be able to provide elemental, molecular and perhaps structural information of the compound(s) responsible of a given chromatographic peak.8 Trace element speciation in the life sciences is rather exploratory at this stage because most of the bioinorganic species present in living organisms are still unknown.61,74 Techniques for identification, confirmation and final quantification of the newly discovered metal-biomolecules are urgently needed nowadays.72 Moreover, validation of speciation results is also increasingly demanded.67 The use of complementary separation methods, orthogonal atomic detectors and molecular structure spectroscopic techniques (so far available only in separate instruments) for each sought species offers at present the more attractive route to tackle speciation problems posed to bio-inorganic analytical chemistry,61,66,68 a real challenge to present atomic spectroscopists. In any case, it should be pointed out that much of the future of speciation analysis will rely on robust, simple and automated sample preparation procedures. Here the exceptional power of FIA strategies62 and flow analysis with atomic detectors75 could play a very important role.
6 Concluding remarks
The evolution of elemental analysis using atomic spectrometry has been spectacular during the last half a century. Of course, fifty years ago multielemental atomic emission techniques were established. However, their sensitivity, selectivity, matrix-free or user-friendly features were rather limited (e.g., one may think about arc spectrography analysis).Both, expected evolution and less expected plasmas and mass detection revolution have brought about a dramatic development of atomic spectrometric techniques, which are able to offer today:
1 Extremely low detection limits, in the ng L−1 range, with the prospect of obtaining in the near future ‘single atom detection’.76
2 Specific detection (i.e., element/isotope selectivity higher than other analytical techniques).
3 Capability, with the adequate accessories for sample introduction, for direct analysis of solids, liquids and gases.
4 Speed of multielement analysis unthinkable a decade ago because of the integration of spectrometer and computer.
5 A wide variety of on line ‘chemistry’ possibilities, in order to enhance the performance of the instrument, are afforded via flow analysis with atomic detectors.8
6 Complex spectrometers more and more computerized are available in the market allowing their handling and control by relatively untrained operators (see Fig. 6 to visualize present trend to integrate chemistry, physics and mathematics knowledge into modern spectrometers).
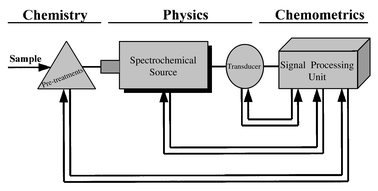 | ||
| Fig. 6 The ‘integrated’ spectrometer with feedback signal processing. | ||
7 Alternative detection systems offer huge capabilities for extracting analytical information. Particularly via ion mass analysers, a bridge between the well developed ‘organic’ MS and the developing atomic (‘inorganic’) MS is expected. Organometallic and metal-biomolecules speciation studies could contribute to build the needed bridge between the two fields of research.
8 Atomic spectrometric detectors are expanding their scope: from total trace element determinations to speciation analysis,60–62 from conventional elemental to isotopic determinations,77 from bulk analyses to localized (compartmental) analyses of the trace elements in the sample,78 or even direct characterization of local constituents and species at the surface of solids.79
9 Miniaturization of machines and instruments seems to be a revolution taking place just now.80 The idea of ‘labs on a chip’ for chromatography is now a reality and can be extended to analytical atomic spectrometry in the near future using ‘microplasmas’81 as miniaturized element-specific detectors which could pave the way to developing simple and chip elemental or atomic chemical sensors82 for real-time, in-situ monitoring of the elements of the periodic table in particular samples (matrices).
Evolution is the mechanism producing the diversity of life83 and natural selection dictates the successful biological species in the long run. Similarly, atomic spectrometry has been in continuous evolution with two important recent revolutions in such natural processes: concerning methodology it appears now that slogans like ‘don’t do it with light, do it with ions’84 could become commonplace in the near future. Moreover, the very essence of analytical atomic spectroscopy could be suffering a sort of metamorphosis with slogans like ‘to look for atoms is useless or even misleading, go for species/compounds’.
Let me conclude by saying that, fortunately for analytical chemists, analytical problems are today so varied, numerous and diverse that it is unwise to think of a single technique, strategy or policy to solve all of them.
In any case, it will be really exciting to follow future analytical atomic spectrometry developments because apparently the end of this century and millennium seems to mark also the end of old concepts (photons for atomic information). This metamorphosis we are witnessing could give birth to new measuring forms and a much wider scope (ions and molecular information) for this rather traditional field.
References
- J. B. Dawson, J. Anal. At. Spectrom., 1991, 6, 93 RSC.
- J. M. Mermet, Les Enjeux Economiques de l’Analyse, Invited lecture at the University of Pau, France, April, 1998. Search PubMed.
- R. S. Houk, V. A. Fassel, G. D. Flesh, H. J. Svec, A. L. Gray and C. E. Taylor, Anal. Chem., 1980, 52, 2283 CrossRef CAS.
- A. Montaser, Appl. Spectrosc., 1998, 52, 406A Search PubMed.
- A. Sanz-Medel, Tec. Laboratorio, 1997, 218, 17 Search PubMed.
- B. R. Levin, M. Lipsitch and S. Bonhoefler, Science, 1999, 283, 806 CrossRef CAS.
- A. Sanz-Medel, Analyst, 1995, 120, 799 RSC.
- O. F. X. Donard and R. Lobinski, J. Anal. At. Spectrom., 1996, 11, 871 RSC.
- A. Sanz-Medel, Mikrochim. Acta (Wien), 1991, II, 265 Search PubMed.
- J. J. Sullivan and B. D. Quimby, Anal. Chem., 1990, 62, 1034 CrossRef CAS.
- J. A. C. Broekaert, Anal. Proc., 1990, 27, 336 RSC.
- J. Kawai, J. Anal. At. Spectrom., 1999, 14, 455 RSC.
- P. Duncumb, J. Anal. At. Spectrom., 1999, 14, 357 RSC.
- A. T. Ellis, P. J. Potts, M. Holmes, G. J. Oliver, Ch. Streli and P. Wobrauschek, J. Anal. At. Spectrom., 1997, 12, 473R Search PubMed.
- R. Klockenkämper and A. von Bohlen, X-Ray Spectrom., 1996, 25, 156 CrossRef.
- F. M. Pennebaker, D. A. Jones, C. A. Gresharn, R. M. Williams, R. E. Simon, M. F. Schapper and M. Bonner Denton, J. Anal. At. Spectrom., 1998, 13, 821 RSC.
- T. W. Barnard, M. I. Crockett, J. C. Ivaldi and P. L. Lundberg, Anal. Chem., 1993, 65, 1225 CrossRef CAS.
- T. W. Barnard, M. I. Crockett, J. C. Ivaldi, P. L. Lundberg, D. A. Yates, P. A. Levine and D. J. Sauer, Anal. Chem., 1993, 65, 1231 CrossRef CAS.
- C. Dubuissen, E. Poussel and J. M. Mermet, J. Anal. At. Spectrom., 1997, 12, 281 RSC.
- L. Soudier and J. M. Mermet, Appl. Spectrosc., 1995, 10, 1478 Search PubMed.
- G. M. Hieftje, J. Anal. At. Spectrom., 1992, 7, 69 RSC.
- R. E. Russo, Appl. Spectrosc., 1995, 49, 14A Search PubMed.
- D. L. Tsalev, J. Anal. At. Spectrom., 1999, 14, 147 RSC.
- A. Sanz-Medel, M. R. Fernández de la Campa, E. Blanco and M. L. Fernández-Sánchez, Spectrochim. Acta, Part B, 1999, 54, 251 CrossRef.
- W. Frech, Fresenius’ J. Anal. Chem., 1996, 355, 475 CAS.
- A. Zybin, C. Schnurer-Patschan and K. Niemax, J. Anal. At. Spectrom., 1995, 10, 563 RSC.
- V. Liger, A. Zybin, Y. Kuritsyn and K. Niemax, Spectrochim. Acta, Part B, 1997, 52, 1125 CrossRef.
- J. M. Harnly, J. Anal. At. Spectrom., 1999, 14, 137 RSC.
- U. Kurfürst, in Solid Sample Analysis. Direct and Slurry Analysis Using GF-AAS and ETV-ICP, ed. U. Kurfürst, Springer Verlag, Berlin, (1998), 1–127. Search PubMed.
- J. Bull and H. Wichman, Science, 1998, 281, 1959 CrossRef CAS.
- C. Th. J. Alkemade, Proc. Soc. Anal. Chem., 1973, 10, 130 RSC.
- A. Montaser, Inductively Coupled Plasma Mass Spectrometry, Wiley-VCH, New York, 1998. Search PubMed.
- J. R. Moody and M. S. Epstein, Spectrochim. Acta, Part B, 1991, 46, 1571 CrossRef.
- K. G. Heumann, S. M. Gallus, G. Radlinger and J. Vogl, J. Anal. At. Spectrom., 1998, 13, 1001 RSC.
- H. M. Kingston, D. Huo, Y. Lu and S. Chalk, Spectrochim. Acta, Part B, 1998, 53, 299 CrossRef.
- A. Sanz-Medel, Spectrochim. Acta, Part B, 1998, 53, 197 CrossRef.
- K. Yokoi, N. W. Alcock and H. H. Sandstead, Biomed. Res. Trace Elem., 1994, 5, 69 Search PubMed.
- M. Patriarca, T. D. B. Lyon, B. McGaw and G. S. Fell, J. Anal. At. Spectrom., 1996, 11, 297 RSC.
- F. Adam, R. Gÿbels and R. Van Grieken, Inorganic Mass Spectrometry, John Wiley and Sons, New York, 1988. Search PubMed.
- R. K. Marcus, Glow Discharge Spectroscopies, Plenum, New York, 1993. Search PubMed.
- C. R. Shick, P. A. De Palma and R. K. Marcus, Anal. Chem., 1996, 68, 2113 CrossRef CAS.
- W. Hang and W. W. Harrison, Anal. Chem., 1997, 69, 4957 CrossRef CAS.
- E. H. Evans and J. J. Giglio, J. Anal. At. Spectrom., 1993, 8, 1 RSC.
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53, 1739 CrossRef.
- J. M. Marchante-Gayón, C. Sariego, J. I. García Alonso and A. Sanz-Medel, Anal. Chim. Acta, (in the press). Search PubMed.
- Z. Du, T. N. Olney and D. J. Douglas, J. Am. Soc. Mass Spectrom., 1997, 8, 1230 CrossRef CAS.
- P. Myers, M. J. Heintz, P. P. Mahoney, G. Li and G. M. Hieftje, Appl. Spectrosc., 1994, 48, 1337 Search PubMed.
- G. M. Hieftje, P. Myers, G. Li, P. P. Mahoney, T. W. Burgoyne, S. J. Ray and J. P. Guzowski, J. Anal. At. Spectrom., 1997, 12, 287 RSC.
- G. C. Eiden, C. J. Barinaga and D. W. Koppenaal, Rapid Commun. Mass Spectrom., 1997, 11, 37 CrossRef CAS.
- C. G. Gill and M. W. Blades, J. Anal. At. Spectrom., 1993, 8, 261 RSC.
- K. E. Milgram, F. M. White, K. L. Goodner, C. H. Watson, D. W. Koppenaal, C. J. Barinaga, B. H. Smith, J. D. Winefordner, A. G. Marshall, R. S. Houk and J. R. Eyler, Anal. Chem., 1997, 69, 3714 CrossRef CAS.
- J. Eyler, E. Milgram and C. Watson, ICP Inf. Newsletter, 1999, 24, 804 Search PubMed.
- A. J. Walder and P. A. Freedman, J. Anal. At. Spectrom., 1992, 7, 571 RSC.
- P. J. Turner, A. N. Eaton, F. Abou-Shakra, J. Speakman, Z. Palauz and P. Brooker, In situ high precision isotope ratios by LA-MC-ICPMS, 6th International Conference on Plasma Source MS, Sept. 1998, Durham, UK. Search PubMed.
- R. E. Sturgeon and R. Guevremont, Anal. Chem., 1997, 69, 2129 CrossRef CAS.
- A. Montaser, Inductively Coupled Plasma Mass Spectrometry, Wiley-VCH, New York, 1998. Search PubMed.
- E. H. Evans, J. J. Giglio, T. M. Castillano and J. A. Caruso, Inductively Coupled and Microwave Induced Plasma Sources for Mass Spectrometry, ed. N. W. Barnett, Royal Society of Chemistry, Cambridge, UK, 1995. Search PubMed.
- R. G. Wilson, F. A. Stevie and C. W. Magee, Secondary Ion Mass Spectrometry, Wiley, New York, 1989. Search PubMed.
- A. Vertes, R. Gijbels and F. Adams, Laser Ionization Mass Analysis, Wiley, New York, 1993. Search PubMed.
- R. Lobinsky, Appl. Spectrosc., 1997, 51, 260A Search PubMed.
- A. Sanz-Medel, M. R. Fernández de la Campa, E. Blanco and M. L. Fernández-Sánchez, Spectrochim. Acta, Part. B, 1999, 54, 274 CrossRef.
- B. Welz, Spectrochim. Acta, Part B, 1998, 53, 169 CrossRef.
- R. M. Harrison and S. Rapsomanikis, Environmental Analysis using Chromatography Interfaced with Atomic Spectroscopy, Horwood, Chichester, 1989. Search PubMed.
- I. K. Krull, Trace Metal Analysis and Speciation, Elsevier, Amsterdam, 1991. Search PubMed.
- A. M. Ure and C. M. Davidson, Chemical Speciation in the Environment, Blackie, London, 1995. Search PubMed.
- S. Caroli, Element Speciation in Bioinorganic Chemistry, vol. 15, Chemical Analysis, Wiley-Interscience, New York, 1996. Search PubMed.
- P. Quevauviller, Mikrochim. Acta, 1996, 123, 3 CAS.
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994. Search PubMed.
- E. H. Evans, W. Pretorius, L. Ebdon and S. Rowland, Anal. Chem., 1994, 66, 3400 CrossRef CAS.
- P. P. Mahoney, J. P. Guzowski-Jr, S. J. Ray and G. M. Hieftje, Appl. Spectrosc., 1997, 51, 1464 Search PubMed.
- M. Belkin, J. W. Waggoner and J. A. Caruso, Anal. Commun., 1998, 35, 281 RSC.
- X. Z. Xiang, C. Y. Ko and H. Y. Guth, Anal. Chem., 1996, 68, 3726 CrossRef CAS.
- G. R. Agnes and G. Horlick, Appl. Spectrosc., 1995, 49, 324 Search PubMed.
- A. Sanz-Medel, Analusis, 1998, 26, 76 Search PubMed.
- A. Sanz-Medel, Flow Analysis with Atomic Spectrometric Detectors, Analytical Spectroscopy Library, vol. 9, Elsevier, 1999. Search PubMed.
- H. Falk, J. Anal. At. Spectrom., 1992, 7, 255 RSC.
- L. Rottmann and K. G. Heumann, Anal. Chem., 1994, 66, 3709 CrossRef CAS.
- J. S. Vogel and K. W. Turteltaub, Nucl. Instrum. Meth. Phys. Res. B, 1994, 92, 445 Search PubMed.
- K. Poels, L. Van Vaeck and R. Gijbels, Anal. Chem., 1998, 70, 504 CrossRef CAS.
- I. Amato, Science, 1998, 282, 402 CrossRef CAS.
- C. Brede, S. Pedersen-Bjergaard, E. Lundanes and T. Greibrokk, Anal. Chem., 1998, 70, 513 CrossRef CAS.
- J. M. Costa, PhD Thesis, Oviedo, 1998..
- B. Hanson, G. Chin, A. Sugden and E. Culotta, Science, 1999, 284, 2105 CrossRef.
- M. W. Blades, Appl. Spectrosc., 1994, 48, 12A Search PubMed.
Footnote |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| This journal is © The Royal Society of Chemistry 2000 |