Determination of cobalt and iron in estuarine and coastal waters using flow injection with chemiluminescence detection†
Vincenzo Cannizzaro, Andrew R. Bowie, Anton Sax‡, Eric P. Achterberg and Paul J. Worsfold*
Department of Environmental Sciences, Plymouth Environmental Research Centre, University of Plymouth, Plymouth, UK PL4 8AA
First published on UnassignedUnassigned7th January 2000
Abstract
Flow injection with chemiluminescence detection (FI-CL) was used to determine cobalt and iron in estuarine and coastal waters. Cobalt(II) was determined by means of a pyrogallol–hydrogen peroxide–sodium hydroxide reaction in the presence of methanol and the surfactant cetyltrimethylammonium bromide (CTAB). With pyrogallol, the sensitivity was enhanced compared with the traditional reagent, gallic acid. The practical limit of detection in sea-water was 5 pM (3s) and there was good agreement with certified values for the sea-water CRMs NASS-5 (0.16 ± 0.01 nM), CASS-3 (0.60 ± 0.09 nM) and SLEW-2 (0.93 ± 0.13 nM). Results for an Irish Sea sample gave good agreement with data obtained using cathodic stripping voltammetry. Iron(II + III) was determined using a luminol reaction with dissolved oxygen as the oxidant. The practical limit of detection was 40 pM (3s) and results from shipboard analysis of the CRM NASS-4 (1.95 ± 0.14 nM) were in good agreement with the certified value (1.88 ± 0.29 nM). Field evaluation of the instrumentation and analytical methods was achieved through a series of local surveys in the Tamar Estuary (UK), from which environmental data are presented.
Introduction
The oceanic concentrations of cobalt are extremely low and it has been suggested that the element may act as a (co-)limiting nutrient for marine phytoplankton.1 Cobalt demonstrates a scavenged-type vertical distribution in the open ocean, typically 18–300 pM in surface waters and 20–50 pM at depth.2 Cobalt concentrations in estuarine and coastal waters are significantly higher. For example, Knauer et al.1 reported 0.85–20 nM in north San Francisco Bay and Achterberg et al. 3 found 140–310 pM in coastal waters near the Wash and Humber Estuaries on the eastern coast of the UK. High concentrations of Co (up to 3.5 nM) have also been reported in hydrothermal plumes4 and the element has been used as a chemical indicator of hydrothermal activity.5 Cobalt is a co-factor in the vitamin B12 complex and is known to accumulate in manganese nodules. It is only toxic to plants and mammals at relatively high concentrations (>17 μM), which are rarely observed in the aquatic environment.6,7Iron has played a key role in oceanographic research over the past decade and is now thought to act as a limiting micronutrient regulating biological productivity in ca. 40% of the world’s oceans.8–10 Consequently, Fe has been intimately linked to the ocean–atmosphere carbon dioxide exchange11 and transitions in climate from glacial to interglacial times.12 Recent advances in analytical and sampling techniques for trace metal determinations have led to an increased understanding of the biogeochemical role of Fe in sea-water, although its distribution in many remote seas and across ocean margins is poorly constrained. Iron has been reported to demonstrate a nutrient-like profile in many regions of the open ocean, with dissolved Fe typically existing at <0.2 nM in surface waters and converging to 0.7–0.8 nM in deep waters.13 However, recent studies suggest that Fe distributions show high temporal and spatial variability through oceanic provinces where atmospheric fluxes are high (e.g., in the Atlantic Ocean14). In coastal and shelf seas, trace metals are delivered laterally by rivers and through diffusion from reductive shelf sediments, resulting in elevated Fe levels (e.g., 1.7 nM in the North Sea15). Estuaries, however, act as a filter, effectively trapping elevated riverine concentrations of metals and leading to large property gradients existing at the land–sea margin.16
Commonly used laboratory techniques for sub-nanomolar Co and Fe determinations include electrothermal atomic absorption spectrometry (ETAAS) following filtration and extractive complexation17 and inductively coupled plasma mass spectrometry (ICP-MS).18 There is a need, however, to develop shipboard analytical methods owing to the problems of sample instability and contamination during transport and storage. ICP-MS and ETAAS are impractical for shipboard use because the instrumentation is bulky, expensive and sensitive to the ship’s vibrations, and the preferred methods for Co and Fe are based on voltammetry,15,19,20 catalytic spectrophotometry21 and flow injection with liquid-phase chemiluminescence detection (FI-CL).14,22–30 All require preconcentration utilising solvent extraction, electrochemical deposition of a metal–ligand complex or chelating resin column separation. Advantages of the FI-CL approach are the ability to perform in-line matrix removal and preconcentration, low-cost detection, wide dynamic range, rapid analysis (seconds), robustness and portability.
In this work, the relative sensitivities of seven polyhydroxy aromatic compounds were evaluated for the FI-CL determination of Co(II). A reaction based on the oxidation of pyrogallol was optimised to permit a limit of detection of 5 pM in sea-water. A modified FI-CL method for the determination of Fe(II + III) in estuarine waters, based on the luminol chemistry, is also reported. An inert tangential flow device is described for filtration of waters containing high suspended particulate matter. The instrumentation was successfully deployed along an axial transect and during two tidal cycles in the Tamar Estuary (UK) and environmental data from these surveys for Co and Fe are presented.
Experimental
Reagents
All plasticware was cleaned by first soaking in hot 5% v/v micro-detergent (DECON; Merck BDH, Poole, Dorset, UK) for 24 h, followed by 1 week in 6 M hydrochloric acid (AnalaR; Merck BDH) and 1 week in 2 M nitric acid (AnalaR; Merck BDH). Laboratory ware was thoroughly rinsed with ultra-high purity (UHP) water (18.2 Ωm cm−1; Elgastat Maxima). All reagents and standards were of analytical-reagent grade, supplied by Merck BDH, unless stated otherwise, and were used as received; solutions were prepared in UHP water. In order to reduce airborne contamination, sample handling in the laboratory was carried out in a class-100 laminar flow hood (Bassaire Model A3VB) and for field studies, work was performed within a specially constructed sealed acrylic hood.Purified acids were used throughout. Hydrochloric acid (9 M) and acetic acid (17.5 M) were purified by single distillation of the analytical-reagent grade acids in a quartz-finger, sub-boiling distillation apparatus to provide Q-HCl and Q-acetic acid, respectively. An ammonia solution (ca. 6 M) was purified using isothermal distillation and nitric acid (15.5 M) was used as received. Standard solutions of Co(II) and Fe(II) were prepared daily in 0.01 M Q-HCl. Samples were diluted with UHP water as necessary.
For the determination of Co, methanol, ethanol, propan-2-ol, propan-1-ol, acetonitrile (HPLC grade; Rathburn Chemicals, Walkerburn, UK), ethylene glycol (puriss. p.a.; Fluka, Buchs, Switzerland), pyrogallol (1,2,3-trihydroxybenzene, ACS grade; Aldrich, Milwaukee, WI, USA), gallic acid (3,4,5-trihydroxybenzoic acid), hydroquinone (benzene-1,4-diol), resorcinol (benzene-1,3-diol), catechol (benzene-1,2-diol), 2,3,4- trihydroxybenzoic acid (all purum ACS; Fluka), 1,3,4- trihydroxybenzene (99%; Acros, Geel, Belgium) and cetyltrimethylammonium bromide (CTAB; Microselect 99%; Fluka) were all used without further purification. The optimum pyrogallol reagent solution was prepared by dissolving 6.3055 g of pyrogallol and 9.1115 g of CTAB in UHP water, adding 113.4 g of 30% v/v hydrogen peroxide (Merck BDH) and diluting to 1 l with UHP water. Silicone oil (Lancaster, Lancs., UK) was used in the thermostating bath and was stable up to 180 °C.
For the determination of Fe(II + III), a working standard reagent solution of luminol (1 × 10−5 M) was prepared 24 h in advance by dilution of a 0.01 M stock standard solution with 0.10 M Na2CO3 and adjusting to pH 12.2 with ca. 2 M NaOH solution. This reagent was passed through a Chelex-100 chelating resin column prior to use in order to reduce the baseline noise generated by trace Fe impurities in the reagents. A working standard sample buffer solution (0.4 M) was prepared from a 2 M stock standard solution of ammonium acetate and adjusted to pH 5.5 with Q-acetic acid. In the FI manifold, this buffer was cleaned in-line using two 8-hydroxyquinoline columns in series. An Fe(III) reducing agent consisting of 100 μM Na2SO3 (extra pure; Merck BDH) was prepared from a 0.4 M stock standard solution which had been pre-cleaned by passing through two sequential 8-hydroxyquinoline columns just prior to use. Aliquots of 2.5 μl were added per millilitre of acidified sea-water to achieve a final sulfite concentration of 100 μM in the sample. The reducing reagent was allowed to react with the acidified sample for at least 8 h. The eluent solution (0.09 M) was prepared by diluting 5 ml of Q-HCl (9 M) to 500 ml with UHP water. An acid wash solution (0.6 M HCl to 0.16 M HNO3) was prepared by diluting 7 ml of purified HCl (9 M) and 1 ml of HNO3 (15.5 M) to 100 ml with UHP water.
Instrumentation
The generic FI-CL instrumentation allowed the selective determination of either Co(II) or Fe(II + III) with only minor modifications to the manifold. The configuration used for the Co(II) determination is shown in Fig. 1. Two peristaltic pumps (Gilson Minipuls 3, Villiers-le-Bel, France) were used to deliver the sample and buffer, UHP water, eluent and reagents. All manifold tubing was PTFE (0.75 mm id (Fisher Scientific, Loughborough, Leics., UK) except for the peristaltic pump tubing, which was Tygon (Elkay, Hants, UK). A six-port PTFE rotary injection valve [Rheodyne (Cotati, CA, USA), Model 5020] was used for sample introduction. The flow cell was a quartz glass spiral (1.1 mm id, 130 μl internal volume) positioned in front of a mirror in a sealed housing. The detection system consisted of an end-window photomultiplier tube (Thorn EMI, 9798QA, Ruislip, Middlesex, UK) contained in a μ-metal shield for magnetic insulation (M552D), a built-in current-to-voltage amplifier (C634), an ambient temperature shielded housing (B2F/RFI) and a 1.165 kV power supply (Thorn EMI, PM28B). The amplifier was supplied with 15 V from an independent power supply (BBH Products, Leeds, UK). The PMT took 3 h to stabilise and then remained powered up. The reagent flow created a background CL emission of 0.15 mV with a peak-to-peak noise of 0.1 mV. A flatbed chart recorder (Kipp and Zonen, BD111, Netherlands) was used to record the output.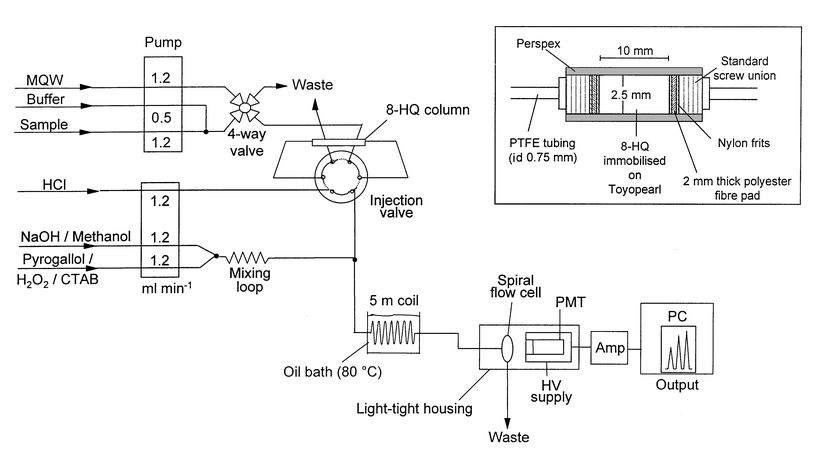 | ||
| Fig. 1 FI-CL manifold used for the determination of Co(II) in estuarine and coastal waters. The inset shows an expanded view of the preconcentration column. | ||
The FI-CL manifold configuration used for the Fe(II + III) determination is shown in Fig. 2. The operating parameters were as described previously,26 except for the incorporation of a 10-port autosampler unit (Valco, Switzerland) which was fully automated using a modified QuickBasic™ software routine. This unit provided improved sample throughput (18 min for triplicate injections of sample plus standard addition) and reduced sample handling.
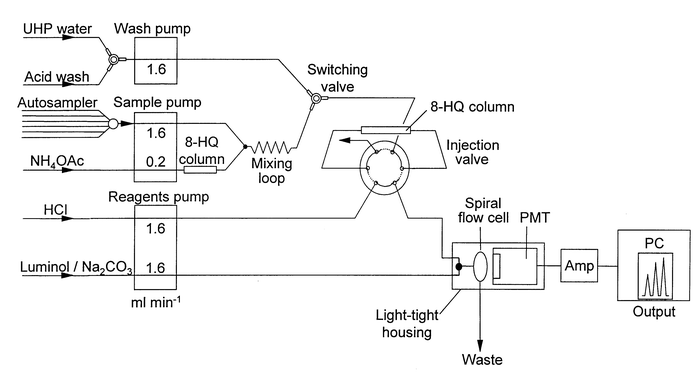 | ||
| Fig. 2 FI-CL manifold used for the determination of Fe(II + III) in estuarine and coastal waters. | ||
Operating procedures
All PTFE flow lines, fittings and connectors of the FI manifold were initially cleaned with 0.5 M Q-HCl and UHP water for at least 2 h. For the determination of Co(II) in sea-water, sample was buffered in-line to pH 5.1 with ammonium acetate (0.1 M) and loaded on to the chelating resin column (see below for details) for 60 s (unless stated otherwise) at a flow rate of 1.2 ml min−1. UHP water was then passed through the column for 30 s to remove the major sea-water cations and anions. The injection valve was switched to the elute position for 60 s and 0.05 M Q-HCl was passed through the column in the reverse direction at a flow rate of 1.2 ml min−1 to elute the Co(II). The eluent stream then merged with the reagent solutions and passed through a 5 m reaction coil (immersed in a heated oil-bath at 80 °C) to the flow cell. The injection valve was then returned to the load position and washed with UHP water for 30 s to remove residual HCl before starting the next load sequence. The load, wash and elution cycle for the Fe(II + III) manifold was as described previously.26Preconcentration
A microcolumn was filled with 8-hydroxyquinoline immobilised on a hydrophilic ethylene glycol–methacrylate copolymer, synthesised according to the method of Landing et al.31 using Toyopearl HW-75F resin (TSK-8HQ, 30–60 μm, fine; Toso-Haas, Anachem, Luton, Beds., UK). The new and robust microcolumn design (Daviron Instruments, Bere Alston, Devon, UK) was constructed from cast acrylic rod and the TSK-8HQ resin sealed inside using standard screw fittings with flanged tubing and nylon frits sandwiching a 2 mm thick polyester fibre pad at each end (shown as the inset in Fig. 1). The column was incorporated in the loop of a six-port rotary injection valve (Rheodyne 5020, HPLC Technology) and cleaned with 0.5 M Q-HCl for 4 h, followed by UHP water for 1 h prior the use.Sampling and filtration
All samples were collected in acid cleaned 250 ml HDPE (high density polyethylene) bottles, which were rinsed three times, filled and placed in sealed polythene bags. For the axial transect, discrete samples were collected by hand at the bow of the research vessel, with the user wearing clean polythene gloves and taking care to minimise contamination. These samples were filtered at sea through an acid cleaned (0.05 M Q-HCl, 24 h) 0.40 μm polycarbonate membrane filter (Nuclepore, Pleasanton, CA, USA) mounted between PTFE supports. During two shore-based 14 h tidal cycle surveys of the Tamar Estuary, samples were fed directly to a remotely powered mobile laboratory using an on-line pumping system.32 A tangential filtration device (Fig. 3) was designed in order to remove suspended particulate matter33 on-line prior to introduction into the FI manifold. All determinations were carried out in near-real time on-board ship or in the field. Samples were diluted with UHP water where necessary to bring them within the linear range of the analytical method.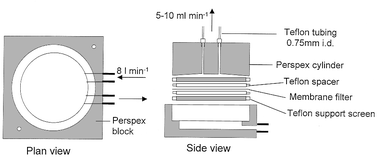 | ||
| Fig. 3 On-line tangential filtration unit for the removal of suspended particulate matter from estuarine samples (modified from Morris et al.33). Sample water was pumped through the lower section of the unit at ca. 8 l min−1, and pulled across a Nuclepore polycarbonate membrane (0.40 μm, 47 mm diameter) mounted in a PTFE sandwich at 5–10 ml min−1. Two output channels allowed sample to be introduced simultaneously into the Co and Fe FI-CL manifolds. All other fittings were chemically inert. | ||
Results and discussion
Optimisation of the Co(II) reaction manifold
The Trautz–Schorigen reaction (TSR)34 involves the oxidation of gallic acid (3,4,5-trihydroxybenzoic acid) by hydrogen peroxide in the presence of a trace metal catalyst to produce CL emission in the visible region. This reaction has been used in an FI-CL manifold for the determination of Co(II) in sea-water23 but other workers have suggested that pyrogallol can be used in place of gallic acid.35 In this work, the CL emission from seven polyhydroxy aromatic compounds was investigated using a 850 pM Co(II) standard solution prepared in 0.01 M Q-HCl. For these experiments, the FI manifold shown in Fig. 1 was modified, with a 200 μl sample loop replacing the preconcentration column and sample buffer line. The relative responses were normalised to gallic acid (100%). Pyrogallol was the most sensitive reagent (140%), with 2,3,4-trihydroxybenzoic acid (14%) and catechol (3%) giving relatively weak responses and resorcinol, hydroquinone and 3,4-hydroxyphenol showing no response. The RSDs (n = 3) were in the range 3–7% for all experiments. Pyrogallol was therefore used for all subsequent experiments. It is known that pyrogallol undergoes rapid auto-oxidation in alkaline solution to give a background CL emission,36 and the reagent solution was therefore prepared fresh each day with UHP water and merged in-line with NaOH as shown in Fig. 1. This solution was found to be stable for at least 24 h at room temperature.In order to maximise sensitivity for the determination of Co(II) using pyrogallol, the effect of key variables (concentration of pyrogallol, CTAB, hydrogen peroxide and methanol and pH) was investigated using the modified FI-CL manifold and an 850 pM Co(II) standard. It is often desirable to use a multivariate procedure, e.g., simplex, to optimise FI manifolds. In this case, however, a univariate approach was preferred because the key variables were all reagent concentrations and it was impractical to prepare the required number of mixed reagent solutions. The profiles for each of the variables (Fig. 4) show that the optimum conditions were 50 mM pyrogallol, 25 mM CTAB, 1.0 M hydrogen peroxide, 20% v/v methanol and a pH of 10.35 (0.15 M NaOH). The general trends were similar to those reported by Sakamoto-Arnold and Johnson23 for the gallic acid reaction except that the methanol and hydrogen peroxide concentrations were significantly higher (20% v/v compared with 4% v/v and 1 M compared with 0.4 M, respectively) and CTAB was used to provide a micellar environment for increased sensitivity.37,38 Using pyrogallol there was no detectable noise with 20% v/v methanol and this is the main factor in the improved sensitivity as compared with gallic acid. This allowed the preconcentration time to be reduced from 4 min to 60 s. The role of methanol is that of an organic modifier which is required to prolong the lifetime of the singlet oxygen produced during the reaction.39 Therefore, the effect of other water-soluble organic solvents (ethanol, ethylene glycol, propan-2-ol, propan-1-ol and acetonitrile) was investigated but the maximum responses (normalised to 100% for methanol) were 62, 38, 24, 23 and 12%, respectively. The fact that acetonitrile (which enhances singlet oxygen lifetime more than methanol40) was not as effective as methanol was probably due to the less favourable polarity of the solvent for the CL reaction. The reaction temperature is also important and immersion of the 5 m reaction coil in an oil-bath at 80 °C gave the best sensitivity without any problems due to the formation of air bubbles in the flow lines.
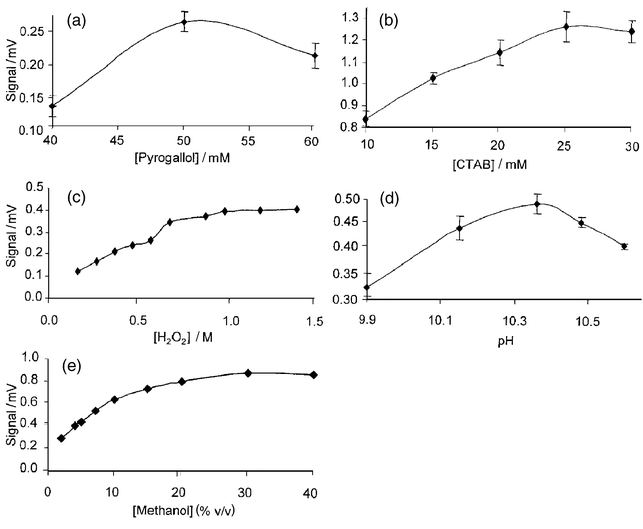 | ||
| Fig. 4 Optimisation of chemical variables for the determination of Co(II): (a) pyrogallol concentration; (b) CTAB concentration; (c) hydrogen peroxide concentration; (d) reaction pH; (e) methanol concentration. Error bars represent 3s (n = 5). | ||
Preconcentration
In order to apply the reaction to the determination of Co(II) in sea-water, it is necessary to introduce a preconcentration and matrix removal step because of the low ambient concentrations of the element in sea-water and the interference from major matrix cations [Ca(II) and Mg(II)].23 The ability of 8-hydroxyquinoline immobilised on Toyopearl (TSK-8HQ) to preconcentrate transition metal ions selectively in the presence of major sea salt cations has been extensively investigated by de Jong et al.27 who reported that 99.92% of Ca(II) and 99.98% of Mg(II) were not retained when 100 ml of sea-water was pumped at 3.9 ml min−1 through a TSK-8HQ microcolumn. It is therefore an excellent chelating resin for use in FI-CL manifolds but the column dimensions and packing of the resin are important for reliable and reproducible performance. In this work, various column lengths and diameters were investigated using a new cast acrylic rod column design (Fig. 1, inset); 2.5 mm was the optimum internal diameter, providing good dispersion characteristics (narrow peaks) with minimum back-pressure, and 10 mm was the optimum length, giving the best dispersion characteristics without any analyte breakthrough. The recovery of Co(II) from the TSK-8HQ column was quantitative over the pH range 4.8–7.8 but decreased rapidly at lower pH, with only 68% recovery at pH 3.8.Interferences
The interference of various transition metal ions [Ag(I), Fe(III), Cu(II), Pb(II), Mn(II), Cd(II)] in the determination of Co(II) by pyrogallol CL was investigated using acidified Atlantic sea-water samples, spiked with Co(II) to give a 250 pM final concentration. Individual metal ions were added at 50 times their typical concentrations found in open ocean sea-water.41 These samples were buffered in-line with ammonium acetate to pH 5.1 and 7.8. Silver(I) was the only element that showed any detectable interference (Table 1), but its concentration in sea-water is typically 0.5–35 pM41 and therefore it is unlikely to interfere in the analysis.| Sample pH | Fe(III) | Cd(II) | Mn(II) | Ag(I) | Cu(II) | Pb(II) |
|---|---|---|---|---|---|---|
| 5.1 | 103 | 102 | 94 | 212 | 101 | 103 |
| 7.8 | 98 | 106 | 97 | 207 | 93 | 100 |
Analytical performance of the cobalt manifold
In most FI-CL reactions with a preconcentration step, the limit of detection is governed by the response from loading and eluting a sample blank. With the pyrogallol reaction, however, the limiting factor is the noise on the baseline from the CL reagents, i.e., there is no detectable signal from acidified UHP water or the ammonium acetate buffer. When using a 60 s sample load time and Co(II) standards prepared in acidified UHP water, the response was linear (r2 consistently >0.999) over the range 5–850 pM with RSDs (n = 3) of 2.1–5.8% and the limit of detection (3 s) was 5 pM. Similar calibration data (offset) were obtained for sea-water but owing to the complex and variable nature of estuarine and coastal waters, a three point standard addition protocol was used for all sample measurements. One sequence of triplicate injections of sample and three standard additions with a 60 s preconcentration (sample load) time took 40 min. The preconcentration time is an important experimental variable with this type of procedure and an 850 pM Co(II) sea-water sample gave a linear response (r2 = 0.9987, n = 4) from 30 to 120 s. Therefore, the preconcentration time can be adjusted to suit the Co(II) concentration of the sample but sample throughput may be a constraint, particularly during shipboard operation.The accuracy of the method was demonstrated by the analysis of three sea-water CRMs, North Atlantic (NASS-4), coastal Atlantic (CASS-3) and St. Lawrence estuarine water (SLEW-2). Excellent agreement with the certified Co(II) concentrations was obtained, as shown in Table 2. Good agreement was also obtained for an Irish sea-water sample (diluted 1 + 1 with UHP water) analysed by FI-CL (r2 = 0.9989 for the standard addition graph) and a CSV method.20
| Sample | FI-CL | Certified value | CSV |
|---|---|---|---|
| NASS-4 | 0.16 ± 0.01 | 0.15 ± 0.02 | — |
| CASS-3 | 0.60 ± 0.09 | 0.68 ± 0.11 | — |
| SLEW-2 | 0.93 ± 0.13 | 0.87 ± 0.21 | — |
| Irish Sea | 0.35 ± 0.02 | — | 0.34 ± 0.01 |
Analytical performance of the iron manifold
The Fe manifold incorporated two technical developments compared with a previously reported system,26 a 10-way automated switching valve and a threaded, cast acrylic rod preconcentration column. The new valve allowed unattended operation during a complete standard addition cycle and improved sample throughput, and the new column resulted in longer operating lifetime and improved reliability. The accuracy was determined on-board ship using NASS-4 and good agreement was obtained between the certificate value (1.88 ± 0.29 nM) and the FI-CL result (1.95±0.14 nM).Tamar field trials
In order to assess the practicality of shipboard operation of the FI-CL analyser and to validate the analytical method for samples containing relatively high trace metal concentrations, three trials were performed locally in the Tamar Estuary (Fig. 5), south-west Devon (UK). Trial I (22nd May 1996) was an axial transect from Plymouth Sound (sea-water end-member) north to Calstock (riverine end-member) and was performed on-board the flat-bottomed fibre-glass research vessel R.V. Tamaris. Trials II and III were land-based studies performed remotely in a mobile laboratory situated on the bank of the Tamar Estuary, near Halton Quay, during two tidal cycles (28th January and 18th March 1997). These surveys were designed to field test the instrumentation and investigate changes in dissolved trace metal concentrations in the estuary. | ||
| Fig. 5 Map of the Tamar Estuary (UK) showing distance in kilometres from Plymouth Sound and the location for the tidal cycle surveys at Halton Quay. | ||
Ancillary shipboard data during trial I (salinity and turbidity) were obtained via an on-board data logging system. During trials II and III, in situ salinity measurements were interpolated from data obtained using a pHOX 52E conductivity meter and dissolved oxygen was measured using a Yellow Springs Industries meter (Model 5739). The concentration of suspended particulate matter (SPM) in samples was determined from the weight of particulate material collected on a pre-weighed 0.45 μm porosity cellulose nitrate filter from a known volume (usually ≡250 ml) of water. The filters were dried overnight at 45 °C and re-weighed on a five decimal place precision balance.
The FI-CL monitor was operated successfully during all three surveys. No major instrumentation problems were experienced despite harsh weather conditions and the only difficulties encountered were de-frosting of the reagents and filtration of estuarine samples containing a high particulate load. Fig. 6(a) shows the results from trial I, mapping the distribution of dissolved Fe, turbidity and salinity against distance from Plymouth Sound, a reference point at the sea-water end of the Tamar Estuary. Fig. 6(b) shows the results for trial II, plotting dissolved Fe, SPM and salinity against sampling time, and Fig. 6(c) presents the data obtained during trial III, plotting dissolved Co, salinity and dissolved oxygen against sampling time. The Fe data are consistent with previously reported studies in the Tamar,42 which observed non-conservative mixing processes with a removal of dissolved Fe due to flocculation of organic matter when salinity increased.43,44 In contrast, Co demonstrates a more uniform distribution across the salinity gradient.
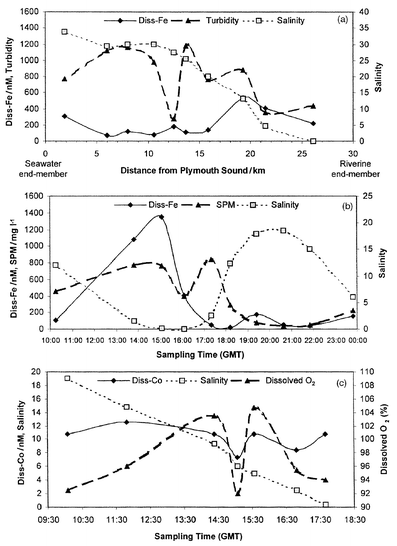 | ||
| Fig. 6 Results from field trials of the FI-CL instrumentation. (a) Dissolved Fe data from an axial transect of the Tamar Estuary; (b) dissolved Fe data from a tidal cycle of the Tamar Estuary; (c) dissolved Co data from a tidal cycle of the Tamar Estuary. | ||
Conclusions
FI-CL manifolds based on the pyrogallol and luminol reactions can be used for the determination of Co and Fe, respectively, in estuarine and coastal waters. The limits of detection (3s) were 5 pM for Co(II) and 40 pM for Fe(II + III). For Co the use of pyrogallol in the presence of CTAB and methanol as an organic modifier significantly improved the signal-to-noise ratio and hence the limit of detection. The procedures were shown to be accurate by obtaining good agreement with certificate values for open ocean, coastal and estuarine CRMs. The FI-CL instrumentation was sufficiently rugged and portable to be deployed on-board ship and on land for monitoring trace metals in estuarine waters and an inert tangential flow filtration device was effective for in-line filtration to remove suspended particulate matter.Acknowledgements
A. B. thanks Tim Fileman for help with the axial transect, Plymouth Marine Laboratory for use of R.V. Tamaris and David Whitworth for assistance during the tidal cycle surveys. The authors thank the EU for funding under the MAST programme (grant number MAS3-CT97-0143, MEMOSEA).References
- G. A. Knauer, J. H. Martin and R. M. Gordon, Nature, 1982, 297, 49 CrossRef CAS.
- J. R. Donat and K. W. Bruland, in Trace Elements in Natural Waters, ed. B. Salbu and E. Steinnes, CRC Press, Boca Raton, FL, 1995, pp. 247–281. Search PubMed.
- E. P. Achterberg, C. Colombo and C. M. G. van den Berg, Cont. Shelf Res., 1999, 19, 537 CrossRef.
- K. L. Von Damm, J. M. Edmont, B. Grant, C. I. Measures, B. Walden and R. F. Weiss, Geochim. Cosmochim. Acta, 1985, 49, 2197 CrossRef CAS.
- H. Sakai, H. Tsubota, T. Nakai, J. Ishibashi, T. Akagi, T. Gamo, B. Tilbrook, G. Igarashi, M. Koreda, K. Shitoashiima, S. Nakamura, K. Fujiioka, M. Watanabe, G. McMurtry, A. Malahoff and M. Ozima, Geochem. J., 1987, 21, 1134 Search PubMed.
- D. R. Williams, The Metals of Life. The Solution Chemistry of Metal Ions in Biological Systems, Van Nostrand Reinhold, New York, 1971. Search PubMed.
- G. N. Schrauzer, in: Metals and their Compounds in the Environment: Occurrence, Analysis, and Biological Relevance, ed. E. Merian, VCH, Weinheim, 1991, pp. 879–892. Search PubMed.
- K. H. Coale, K. S. Johnson, S. E. Fitzwater, R. M. Gordon, S. Tanner, F. P. Chavez, L. Ferioli, C. M. Sakamoto, P. Rogers, F. Millero, P. Steinberg, P. Nightingale, D. Cooper, W. P. Cochlan, M. R. Landry, J. Constantinou, G. Rollwagen, A. Trasvina and R. Kudela, Nature (London), 1996, 383, 495 CrossRef CAS.
- D. A. Hutchins, G. R. Ditullio, Y. Zhang and K. W. Bruland, Limnol. Oceanogr., 1998, 43, 1037 Search PubMed.
- P. W. Boyd, A. Watson, C. S. Law, E. Abraham, T. Trull, R. Murdoch, D. C. E. Bakker, A. R. Bowie, M. Charette, P. Croot, K. Downing, R. Frew, M. Gall, M. Hadfield, J. Hall, M. Harvey, G. Jameson, J. La Roche, M. Liddicoat, R. Ling, M. Maldonado, R. M. McKay, S. Nodder, S. Pickmere, R. Pridmore, S. Rintoul, K. Safi, P. Sutton, R. Strzepek, K. Tanneberger, S. Turner, A. Waite and J. Zeldis, Nature (London), submitted. Search PubMed.
- D. J. Cooper, A. J. Watson and P. D. Nightingale, Nature (London), 1996, 383, 511 CrossRef CAS.
- J. H. Martin, Paleoceanography, 1990, 5, 1 CrossRef.
- K. S. Johnson, R. M. Gordon and K. H. Coale, Mar. Chem., 1997, 57, 137 CrossRef CAS.
- R. T. Powell, D. W. King and W. M. Landing, Mar. Chem., 1995, 50, 13 CrossRef CAS.
- M. Gledhill and C. M. G. van den Berg, Mar. Chem., 1995, 50, 51 CrossRef CAS.
- G. E. Millward, Analyst, 1995, 120, 609 RSC.
- K. W. Bruland, R. P. Franks, G. A. Knauer and J. H. Martin, Anal. Chim. Acta, 1979, 105, 223 CrossRef CAS.
- J. F. Wu and E. A. Boyle, Anal. Chim. Acta, 1998, 367, 183 CrossRef CAS.
- E. L. Rue and K. W. Bruland, Mar. Chem., 1995, 50, 117 CrossRef CAS.
- M. Vega and C. M. G. van den Berg, Anal. Chem., 1997, 69, 874 CrossRef CAS.
- C. I. Measures, J. Yuan and J. A. Resing, Mar. Chem., 1995, 50, 3 CrossRef CAS.
- K. Isshiki and E. Nakayama, Talanta, 1987, 34, 277 CrossRef CAS.
- C. M. Sakamoto-Arnold and K. S. Johnson, Anal. Chem., 1987, 59, 1789 CrossRef CAS.
- T. Yamane and K. Watanabe, Anal. Chim. Acta, 1988, 207, 331 CrossRef CAS.
- H. Obata, H. Karatani and E. Nakayama, Anal. Chem., 1993, 65, 1524 CrossRef CAS.
- A. R. Bowie, E. P. Achterberg, R. F. C. Mantoura and P. J. Worsfold, Anal. Chim. Acta, 1998, 361, 189 CrossRef CAS.
- J. T. M. de Jong, J. den Das, U. Bathmann, M. H. C. Stoll, G. Kattner, R. F. Nolting and H. J. W. de Baar, Anal. Chim. Acta, 1998, 377, 113 CrossRef CAS.
- Y. Masaaki, T. Komatsu, S. Nakahara and S. Suzuki, Anal. Chim. Acta, 1983, 155, 259 CrossRef.
- S. Nakahara, M. Yamada and S. Suzuki, Anal. Chim. Acta, 1982, 141, 255 CrossRef CAS.
- S. Hirata, Y. Hashimoto, M. Aihara and G. V. Mallika, Fresenius’ J. Anal. Chem., 1996, 355, 676 CAS.
- W. M. Landing, C. Haraldsson and N. Paxeus, Anal. Chem., 1986, 58, 3031 CrossRef CAS.
- D. J. Whitworth, E. P. Achterberg, M. Nimmo and P. J. Worsfold, Anal. Chim. Acta, 1998, 371, 235 CrossRef CAS.
- A. W. Morris, R. J. M. Howland and A. J. Bale, Estuarine Coastal Mar. Sci., 1978, 6, 105 Search PubMed.
- M. Trautz and P. Schorigin, Z. Wiss. Photogr. Photophys. Photochem., 1905, 121, 3 Search PubMed.
- R. J. Miller and J. D. Ingle, Talanta, 1982, 29, 303 CrossRef CAS.
- G. B. Meluzova, L. A. Belova and V. G. Voronkov, Russ. J. Phys. Chem., 1970, 44, 744 Search PubMed.
- A. Safavi and M. R. Baezzat, Anal. Chim. Acta, 1998, 368, 113 CrossRef CAS.
- Z. H. Xie, F. Zhang and Y. S. Pan, Analyst, 1998, 123, 273 RSC.
- J. Slawinski, Photochem. Photobiol., 1971, 13, 489 Search PubMed.
- C. L. Shellum and J. W. Birks, Chemiluminescence and Photochemical Reaction Detection in Chromatography, ed. J. W. Birks, VCH, Weinheim, 1989, p. 236. Search PubMed.
- R. Chester, Marine Geochemistry, Unwin Hyman, London, 1990. Search PubMed.
- A. W. Morris, A. J. Bale, R. J. M. Howland, G. E. Millward, D. R. Ackroyd, D. H. Loring and R. T. T. Rantala, Water Sci. Technol., 1986, 18, 111 CAS.
- E. R. Sholkovitz, Earth. Planet. Sci. Lett., 1978, 41, 77 CrossRef CAS.
- L. L’Herroux, S. M. Le Roux and P. Appriou, Mar. Pollut. Bull., 1998, 36, 56 CrossRef CAS.
Footnotes |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| ‡ Present address: IFA-Tulln, Analytikzentrum, Konrad Lorenz Strasse 20, A-3430 Tulln, Austria. |
| This journal is © The Royal Society of Chemistry 2000 |