Determination of carbon-14 levels in heavy water and groundwaters†‡
F. Caron*, J. Sutton, M. L. Benz and M. K. Haas
Atomic Energy of Canada Limited, Environmental Research Branch, Chalk River, ON, Canada K0J 1J0. E-mail: caronf@aecl.ca
First published on UnassignedUnassigned7th January 2000
Abstract
The radionuclide carbon-14 is a by-product of nuclear reactors, produced predominantly by the neutron activation of 17O of the water molecule. The chemical form of carbon-14 in heavy water is bicarbonate (D14CO3−), and the total inorganic carbon levels (TIC) are expected to be 20–30 μg L−1. A technique was developed primarily for 14C determination in reactor heavy water (ultra-pure water containing up to ≡1012 Bq L−1 of tritiated water), and is also applicable to analysis of contaminated groundwater. The method consists in acidifying the sample to convert the bicarbonate into CO2, which is swept by a stream of nitrogen to an absorbent NaOH solution. An aliquot of this NaOH solution is measured by liquid scintillation for counting 14C. Isolation of the 14C from the reactor water matrix was excellent, as tritium removal factors of up to 109 were obtained. The recoveries of simulated 14C solutions were 94.2 ± 6% (n = 61) for five analysts of various levels of experience. The variations in the recoveries followed a non-systematic random pattern, and were independent of the analyst. The 14C concentrations in reactor water sampled during operation were in the range 0.1–2 MBq L−1 with a typical variability of ±5% or better among replicates, while the reactor water samples obtained during shutdown and samples of groundwater near a waste management area had values ranging from ≡100 to 1800 Bq L−1, but with a higher variability. The detection and determination limits were 47 and 342 Bq L−1, respectively.
1 Introduction and background
The radionuclide carbon-14 is a by-product of all nuclear reactors, and is produced predominantly by the neutron activation of 17O of water molecules.1,2 In heavy water reactors such as CANDU® reactors (CANDU® is a registered trademark of Atomic Energy of Canada Limited), the major portion of the 14C produced originates from the moderator. The formation of 14CO2 is favored, hence the predominant chemical form of 14C is as bicarbonate (D14CO3−) at neutral moderator water pH (or pD in heavy water). The levels of D14CO3− present in the moderator depend upon the production term, i.e., reactor power output, and the removal term, i.e., the purification rate, which is controlled by ion-exchange columns used for general purification. A potential diagnostic of the performance of the purification system is the 14C level in the moderator.Conceptually, measurement of 14C of moderator water is straightforward. The moderator consists of ultra-pure water, depleted of ions except for those added voluntarily for reactivity control or shutdown. A major complication arises from the presence of tritiated heavy water (predominantly as 2H3HO or DTO) which is present at high levels [≡0.4 × 106–1.5 × 106 MBq L−1 or 10–40 Ci L−1], and originates from the neutron activation of the deuterium atom of the heavy water. The levels of 14C expected in the moderator of CANDU® reactors are 0.05–0.1 MBq L−1 or ≡5–6 orders of magnitude lower than DTO. Both tritium and 14C are β-radiation emitters and direct counting of moderator water for 14C by liquid scintillation counting (LSC) is nearly impossible, as the signal due to tritium dominates over that of other β-emitters, even if the energies of the other β-emitters do not overlap with tritium. To overcome this problem, a procedure was developed, based on acidifying the sample to release the dissolved 14CO2 as gas, stripping the CO2 from the tritiated water matrix and collecting the CO2 in a base [NaOH or Carbosorb™ (a product from Packard Instruments, Meriden, CT, USA, consisting of an organic amine; the nominal CO2 absorption capacity is 4.8 mmol mL−1)].3 An aliquot of the base or the whole absorbent is mixed with a liquid scintillation cocktail and analyzed using a liquid scintillation counter. In the past, various analysts have used different conditions (volumes of solution, absorbents, acids, hardware designs and different protocols on LSC instruments), and the results have varied between analysts. The variations were potentially caused by system leaks, incomplete or inconsistent stripping of the 14CO2 and/or similar aspects related to differences in the acidification/ absorption protocols. The results presented in this paper from several analysts show the consistency of the method, the removal of interferences from the matrix and that it can be used successfully by analysts of various degrees of experience.
2 Warning and safety precautions
This procedure is for the determination of 14C (present as bicarbonate or carbonate) in small volumes (<100 mL) of tritiated water. The potential hazards are 14C and 3H (tritium or DTO), strong acids and bases used in titrations and magnesium perchlorate, used as a drying agent. The analysts must have proper training for handling of radioactivity and the laboratory must be approved for handling the expected levels of radioactivity. Radioactive wastes must be disposed of according to approved procedures.3 Methodology
3.1 System parameters and description
A drawing showing the extraction line is shown in Fig. 1. The main parts of the system consist of a supply of inert gas (nitrogen), a reaction flask, a drying trap and an alkaline trap (bubbler).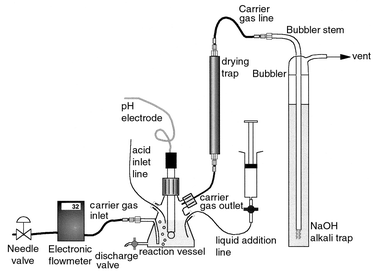 | ||
| Fig. 1 Description of the acid stripping line. | ||
The sweep gas used was nitrogen (zero grade), passed through a soda-lime trap to remove CO2 impurities. An electronic flow meter (Omega Engineering, Stamford, CT, USA) with a separate needle valve allowed precise fine tuning of the gas flow rate to the reaction vessel. The reaction vessel consisted of a 125 mL Erlenmeyer flask, modified with six penetrations. The discharge valve was a standard Pyrex brand, straight bore, PTFE plug stopcock. The carrier gas inlet was made out of an exact, 0.25 in (≡6 mm) glass tube to fit a Swagelok™ PTFE fitting, with the tube running inside the vessel to avoid spillage when filling with liquid. The other inlet lines were from AceTM glass, with rubber septa or O-rings, to make gas-tight connections to the addition lines. The drying trap consisted of a borosilicate glass tube (30 cm × 10 mm id), with smaller Labline™ screw-on inlet and outlet tubes to fit 1/16 in (2 mm) PTFE tubing (Ace Glass). The alkaline trap (bubbler) was made of a 50 cm × 20 mm od tubing to hold 140 mL of alkaline solution. The bubbler was equipped with a fritted tip (porosity C) and was vented to a fume hood.
The system was designed to minimize leaks, thus ensuring maximum 14C recoveries, and minimizing evaporation and potential contact with tritiated water. The pH electrode and the acid inlet line were fed to the reaction vessel by a Tanager (Ancaster, ON, Canada) Model 8901 autotitrator. This was an important component of the system, as the pH of the solution had to be decreased gradually to avoid fast de-gassing. The liquid addition line was fitted to a Luer stopcock for adding various solutions without opening the reaction vessel. The solution in the bubbler was 2 M NaOH. The drying agent consisted of analytical-reagent grade magnesium perchlorate, 4–20 mesh (Fisher Chemicals, Fairlawn, NJ; UN-1475), loosely packed in the trap to minimize the pressure drop. Inconsistent 14C recoveries were obtained when the drying agent was silica gel.
Some of the items of the hardware are not critical, as follows. Flow meter: a ball rotameter or a soap bubble flow meter can be used. The soap bubble flow meter should be added at the outlet of the bubbler, whereas the ball rotameter could be placed at the same location as the current design. Acid delivery system: a syringe pump can be used instead of an automatic titrator. The acidification should be performed over a period similar to the current titration, ≡30–40 min with an acid of moderate concentration (we used 0.5 M). The use of a pH electrode is recommended for consistency, as opposed to using a pH color indicator. Alkaline bubbler: the key item in the bubbler is the fritted tip. The amount of solution that we used (≡140 mL) featured a long travel path (≡40 cm). The exact concentration of NaOH should not be significant and, although we used 2 M NaOH throughout our work, we tested several solutions from 1 to 4 M without apparent differences.
3.2 Samples and standards
The moderator water samples were withdrawn by operators at the station into 125 mL borosilicate glass bottles (Qorpak; Fisher Scientific, Pittsburgh, PA, USA, Cat. No 03-326-1D), with a PTFE (0.25 mm thick) liner, cut manually to fit inside the cap. The bottles were pre-filled with 100 μL of saturated NaOH solution (19.4 M) to preserve the samples at a high pH. The bottles were filled to the top, without headspace. The samples were stored at ambient room temperature and were analyzed ≡30–180 d after their collection. The groundwater samples were collected from a monitoring well near a waste management area at Chalk River Laboratoreis (CRL), and stored in a 1 L Nalgene bottle, along with 1 mL of saturated NaOH solution. The samples were also filled and capped with no headspace, and were preserved in a refrigerator prior to analysis.Standards consisted of Na214CO3 (≡50000 Bq mL−1), prepared by diluting a primary NaH14CO3 solution (50 μCi in a 1 mL sterile ampule; New England Nuclear, Life Sciences, Boston, MA, USA, Cat. No. NEC-0865) in 2 M NaOH solution. Aliquots of our standard were analyzed repeatedly by LSC on several instruments, and compared with a NIST-traceable standard (EG&G Wallac, Gaithersburg, MD, USA) for accuracy. We used glass scintillation vials and Hionic Fluor cocktail (Packard Instruments, Meriden, CT, USA), in a volume ratio of 1∶9 (mL∶mL 2 M NaOH to cocktail). All solutions mixed with the cocktail were 2 M NaOH, or brought to 2 M NaOH to minimize matrix effect differences during counting. All the samples and standards were shaken and allowed to stand for at least 1 h prior to counting, to allow chemiluminescence to decrease (the chemiluminescence should be negligible after 20 min, according to the manufacturer). We tested a waiting time of ≡16 h (overnight) without noticeable difference. Each sample or standard was counted three times, which contributed to decreasing the noise due to chemiluminescence, if significant.
3.3 Procedure for 14C stripping
After filling the drying trap and the alkaline bubbler, the tubing connections were attached between the reaction vessel, the trap and the bubbler. The carrier gas inlet line (without flow), the acid inlet and liquid addition lines were also attached and the discharge valve was kept closed. A magnetic stirrer, 2 mL of 2 M KNO3 solution (for ionic conductivity) and 2 mL of 0.7 M Na2CO3 carrier solution were added through the pH electrode opening of the reaction vessel. The pH electrode was then inserted and secured (gas-tight). Using a 60 mL disposable syringe, 30 mL of heavy water were withdrawn from the sample bottle and the syringe was carefully attached to the liquid addition line and pushed into the reaction vessel through the line. Using a different syringe, a slightly alkaline de-ionized water (DIW) solution (prepared by adding a few drops of saturated NaOH solution to DIW) was carefully added to bring the level of solution up to the pH electrode junction.The carrier gas was then opened at a flow rate of 38 ± 2 mL min−1, and the connections were checked for leaks with a soap solution. The magnetic stirrer was turned on and the titration was started only after successful completion of these checks. Titration gradually decreased the pH to 2 using 0.5 M HCl, over a 30–40 min period. At the end of the titration, a syringe filled with 0.1 M HCl was attached to the liquid addition line and 10–30 mL of acid were added to decrease the volume of headspace in the reaction vessel to a minimum. The system was allowed to purge for an additional 30 min. The same procedure was used for process blanks (DIW only) and process standards (100 μL of Na214CO3 solution, ≡50000 Bq mL−1, in DIW) for recovery checks.
At the end of the acid-stripping procedure, the NaOH solution in the bubbler was recovered first (to avoid the potential pick-up of airborne DTO from the next steps) and poured into a high density polyethylene bottle, which was capped immediately. The reaction vessel was then emptied and its contents were collected for disposal or recovery (heavy water). The 14C sample in the NaOH solution is stable indefinitely.
The groundwater samples were processed similarly to above, except that the flow rate of carrier gas was 30 mL min−1.
3.4 Counting procedure
The samples were counted in three cycles of 10 min each. The counter and the Na214CO3 solution (50000 Bq mL−1) were checked periodically against NIST-traceable 14C and blank solutions (traceability provided by Wallac), and a 2822 dpm mL−1 (47.0 Bq mL−1) NIST-traceable secondary standard (solution prepared on October 6, 1996, from [14C]hexadecane, NIST SRM 4222C). The latter was also used in comparisons with other instruments at CRL. The comparisons with the other instruments suggested that the Wallac 1414 instrument could have a ≡0–3% low bias, but the results were still within instrument and standards specifications, and no correction for LSC accuracy was considered to be needed.
4 Results and discussion
4.1 Precision and discrimination between 14C and 3H
The original samples of heavy water contain known, relatively high levels of tritiated water and unknown amounts of 14C. The 14C analyses of reactor water are shown in Table 1. With a few exceptions, the typical precision for samples having 0.1 MBq L−1 or more was ≡5% or better. Most of the variations could be explained by statistical variations in the efficiencies of the acid-stripping process. On the other hand, the results for which the 14C concentrations were below 0.002 MBq L−1 were limited by the precision of LSC analysis. The process blanks, reported as a percentage of the 14C found in the same set of runs, were usually less than 1%, except for the low 14C solutions. This indicates that memory effects and cross-contamination between samples are negligible.| Sample | 14C concentration in original 30 mL sample MBq L−1 | RSD (%) | Process blank (% of 14C recovered) | H concentration in the 30 mL sample/105 MBq L−1 | Reported 3H in bubbler (total)/Bqa |
|---|---|---|---|---|---|
| a Value reported by the LSC instrument. This signal may not be for tritium (see text).b Samples not analyzed; the values given on the shipping manifests are reported. | |||||
| A1I | 1.3 | 4.3 | 0.5 | 7.4 | 337 |
| A2I | 3.8 | 3.7 | 0.6 | 10.0b | 3069 |
| A3O | 0.023 | 20.0 | 1.1 | 7.2 | 7 |
| A4I | 3.3 | 3.1 | 0.2 | 16 | 1593 |
| B1I | 0.17 | 12.9 | 0.9 | 3.8 | 81 |
| B2I | 1.7 | 5.6 | 0.6 | 3.7b | 613 |
| B3I | 1.8 | 4.2 | 0.4 | 3.0b | 1372 |
| B4I | 3.3 | 4.6 | 0.5 | 3.7 | 1617 |
| C1I | 0.14 | 2.2 | 0.3 | 19.0 | 75 |
| D1I | 0.0016 | 15.0 | 16.0 | 7.5 | 33 |
| D1O | 0.0008 | 25.9 | 33.0 | 6.1 | 24 |
| D2I | 0.0012 | 3.7 | <0.1 | 5.2 | 18 |
| D3I | 3.7 | 4.3 | 0.6 | 3.6 | 2071 |
| D4O | 4.2 | 4.9 | 0.7 | 5.7 | 3132 |
| D5I | 4.2 | 2.7 | 0.8 | 4.6 | 2634 |
| D5O | 4.0 | 4.7 | 0.7 | 5.2 | 2801 |
| D6I | 3.4 | 4.1 | 0.4 | 4.5 | 1595 |
We obtained a high tritium removal factor [defined as total activity (reaction flask)/total activity (bubbler), where total activity (reaction flask) is concentration (MBq L−1) × 0.030 L of initial sample]. The data in Table 1 show that the 3H signal reported in the bubbler ranged from 7 to ≡3100 Bq total, indicating that the removal factor of the stripping process plus the drying trap is at least 5 × 106–3 × 109. The actual removal factor, however, could be higher because the 3H signals, as reported by the Wallac 1414 instrument, are questionable, as we found a positive correlation between the 14C signal and the 3H signal (see second and last columns in Table 1). The cause(s) of this is (are) not clear; it appears that the 3H signal reported is a 14C signal falsely reported in the 3H energy window. We tested and dismissed other causes: for example, we tested with different batches of Hionic Fluor LSC cocktail, chemiluminesence and interferences due to low-energy γ-emitters produced in reactors, such as 41Ar, 16N, 19O and 17F because of their short half-lives, combined with a 1–6 month period prior to sample analysis. Our carbonate standards containing known amounts of 14C gave a signal in the 3H window, but this signal was inconsistent among individual measurements. Conversely, samples containing a single 3H spike [83000 disintegrations per minute (dpm)] did not give a false positive 14C signal. Hence, the actual 3H signal in our samples is most likely lower than reported, which would indicate a higher removal factor than mentioned above.
Table 2 gives the results for the process standards and their associated process blanks, analyzed using the same procedure as for the reactor water. In all but two cases (see Table 2), no tritiated water was present. The precision, given as relative standard deviation (RSD), was generally similar to that of the actual samples. The presence of tritiated water did not affect the precision.
| Processing date (1998) | Total 14C in spikes and test solutions/Bq | Total 14C recovered/Bq | Precision (RSD) (%) | Process blank (% of 14C recovered) |
|---|---|---|---|---|
| a Process blank not applicable to this batch. | ||||
| 27 Jan | 9623 Bq 14C test solution, prepared Dec. 23, 1997 | 9024 | 5.4 | 0.4 |
| 29 Jan | 9623 Bq 14C test solution, prepared Dec. 23, 1997 + 30.7 MBq 3H | 8935 | 2.5 | 0.7 |
| 3 Feb | 4811 Bq 14C spike in DIW | 4493 | 1.5 | N/Aa |
| 24 Feb | 4811 Bq 14C spike in DIW | 4253 | 3.7 | 0.2 |
| 27 Feb | 4811 Bq 14C spike + 30.7 MBq 3H in DIW | 4531 | 2.2 | 0.1 |
| 7 Apr | 9623 Bq 14C test solution, prepared Dec 23, 1997 | 9210 | 0.6 | 0.2 |
| 28 Apr | 5236 Bq 14C spike in DIW | 5601 | 4.0 | 0.3 |
| 4 Jun | 5172 Bq 14C spike in DIW | 4583 | 8.7 | 0.4 |
| 17 Jun | 5172 Bq 14C spike in DIW | 4395 | 5.2 | 0.1 |
| 23 Sep | 5092 Bq 14C spike in DIW | 4833 | 0.5 | N/Aa |
| 17 Nov | 9623 Bq 14C test solution, prepared Dec. 23, 1997 | 9443 | 1.1 | N/Aa |
Table 3 gives the results for groundwater samples and additional details of some low 14C reactor water. One could note an improved precision when a larger 2 mL aliquot is used in LSC counting at these low levels. The method, as used, has the potential to analyze solutions reaching drinking water limits (200 Bq L−1).4 In comparison, the total carbon corresponding to this value is ≡1.4 × 10−9 g L−1 if it was pure 14C. For solutions (reactors or groundwater) containing μg L−1 to mg L−1 levels of total carbon, the radioactive carbon is a small fraction of the total.
| Sample ID | Volume of bubbler solution counted/mL | Process run ID | Count rate/ dpm | Error (RSD) (%) | Concentration in sample/Bq L−1 | Notes | |
|---|---|---|---|---|---|---|---|
| a Three values from three counting cycles are usually reported. | |||||||
| Groundwater— | |||||||
| C-112 | July | 2 | DEC16R3 | 13.5 | 0.7 | 405 | 2 count vaues reporteda |
| Aug. | 2 | DEC16R4 | 4.5 | 16 | 99 | 2 count values reported | |
| 1 | DEC16R4 | 4.4 | 48 | 193 | 2 count values reported | ||
| Sept. | 2 | DEC16R5 | 9.1 | 24 | 245 | 2 count values reported | |
| C-265 | July | 1 | DEC17R2 | 1.9 | 82 | 90 | |
| 2 | DEC17R2 | 3.1 | 11 | 73 | 2 count values reported | ||
| Aug. | 1 | DEC17R5 | 5.1 | 58 | 197 | ||
| 2 | DEC17R5 | 5.5 | 33 | 102 | |||
| Sept | 1 | DEC17R4 | 17.4 | 23 | 1047 | ||
| 2 | DEC17R4 | 25.2 | 3.3 | 757 | |||
| C-267 | July | 1 | DEC16R2 | 22.7 | 0.9 | 1101 | |
| 2 | DEC16R2 | 38.6 | 5.9 | 943 | |||
| Aug. | 1 | DEC15R1 | 4.3 | 53 | 163 | ||
| 2 | DEC15R1 | 9.7 | 22 | 187 | |||
| Sept | 2 | DEC15R2 | 2.8 | 7.1 | 67 | ||
| Reactor— | |||||||
| D1I | 2 | FEB17R1 | 49.5 | 3.1 | 1860 | ||
| FEB17R2 | 40.2 | 1.8 | 1540 | ||||
| FEB17R3 | 25.5 | 6.1 | 1290 | ||||
| Average | 1563 | ||||||
| D1O | 1 | FEB23R2 | 9.1 | 44 | 676 | ||
| FEB23R3 | 5.1 | 48 | 392 | ||||
| FEB23R4 | 6 | 29 | 460 | ||||
| Average | 566 | ||||||
| 2 | FEB23R2 | 25.8 | 8.6 | 960 | |||
| FEB23R3 | 14.2 | 17.4 | 547 | ||||
| FEB23R4 | 15.9 | 6.8 | 606 | ||||
| Average | 804 | ||||||
| D2I | 1 | MAR4R1 | 15.5 | 13.5 | 1200 | ||
| MAR4R2 | 16.1 | 7.4 | 1230 | ||||
| MAR4R3 | 14.5 | 7.2 | 1130 | ||||
| Average | 1187 | ||||||
4.2 Recoveries and robustness between analysts
Fig. 2 shows the recoveries of 14C process standards for sequential runs and the recoveries among different analysts. The method gave an average recovery of 94.2 ± 6% (n = 61) for the various process standards. The system does not have a bias among analysts. Three of the analysts were experienced (Nos. 1, 2 and 5), whereas the other two analysts were relatively inexperienced.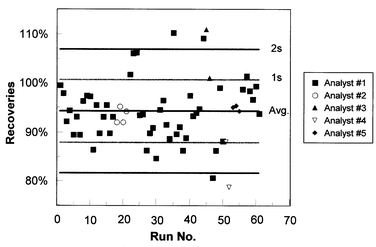 | ||
| Fig. 2 Recoveries of 14C analyses for process standard runs, also showing the recoveries for several analysts with various levels of experience. | ||
The percentage recovery data match reasonably well the pattern of the probability density function, as shown by the frequency distribution of our recoveries compared to the normal distribution (Fig. 3). The median (94%) is very close to the average value for all these points. We conclude, therefore, that the analysis method as presented does not have systematic errors or user bias. The variations of the recoveries can be explained by the normal statistical model.
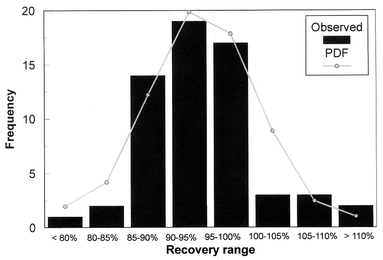 | ||
| Fig. 3 Comparison of the recoveries with a probability density function (PDF). | ||
4.3 Detection and quantification limits
The instrument background noise for the full 14C spectrum is 55.0 ± 2.2 counts per minute (cpm) over a 10 min period. Using the Student’s t-test (two-sample case, large number of data points, i.e., n > 305), the minimum detectable limit of a sample for LSC is 2.5 cpm above the background (95% confidence limit), which corresponds to ≡3 dpm at 90% efficiency for 14C. Using 140 mL as the volume of NaOH solution in the bubbler and a volume of groundwater or reactor water of 75 mL in the reaction vessel, the calculated detection limit is 93 Bq L−1 if 1 mL of the bubbler solution is analyzed by LSC, or 47 Bq L−1 for a 2 mL analysis.A generally accepted value for the quantification limit is when the signal is approximately 10 times the standard deviation associated with the background noise.5 We set this level at 20 cpm, or 22 dpm above the background (at 90% efficiency). Using the same numbers as above, the quantification limit is set at 685 and 342 Bq L−1 for 1 and 2 mL aliquots counted by LSC, respectively.
The results given in Table 3, when revisited, show that only two groundwater samples (C-265, September, 2 mL, and C-267, July) and two reactor water samples (D1I and one of D1O, 2 mL) display a value above the quantification limit of 22 dpm. The values for most of these samples are between the detection and the quantification limits.
4.4 Sample stability
A synthetic 10 kBq solution total (DIW with 100 μL of saturated NaOH and 200 μL of nominal 50000 Bq mL−1 solution) was prepared on December 23, 1997, in a Qorpak bottle with a PTFE-lined cap. Four different bottles were prepared and left at ambient temperature until processing at various times, up to ≡320 d after preparation. The 14C recoveries ranged from 92.8 to 98.2%, which is well within the recoveries found with process standards throughout the study. The samples were stable for this period (≡1 year), which was important, as many of the samples were stored for 3-6 months prior to processing.4.5 Key variables
This work has overcome several fine-tuning problems that were encountered in the past by several different analysts. In principle, the method is simple, but in practice, several aspects of the hardware and the procedure can produce inconsistent results.We used a 0.5 M acid, compared with undiluted or 2–4-fold diluted commercial concentrated acids, as often used previously and in early tests. With stronger acids, a sudden release of CO2 was often clearly visible in the NaOH bubbler, thus increasing the potential for losses through leaks or inefficient contact with the alkaline solution. The use of a titrator for gradual acidification through the S-shaped part of the titration was also a factor that contributed to a controlled release of CO2.
The bubbler had a long contacting path between the impinger and the top of the solution (≡40 cm), and a frit to divide the gas bubbles finely. Tests carried out with ≡10 MBq of 14C in the reaction flask going to two bubblers in series showed no significant amounts of 14C in the second bubbler, hence the 14C recovery is quantitative in the first bubbler (results not shown).
The design of the reaction flask was important. The conical shape of the reaction vessel and the addition of 0.1 M HCl provided a maximum displacement of the headspace to ≡5% of the flask volume. This resulted in a combined flushing time of ≡3.1 min in the flask, which corresponds to a first-order flushing rate of 3.7 × 10−3 s−1. In comparison, the conversion reactions to CO2 were much smaller:6
| H+ + HCO−3 → H2CO3 k1 = 4.7 × 1010 l mol−1 s−1 | (1) |
| H2CO3 → H2O + CO2 k2 = 11.9 s−1 | (2) |
| H+ + HCO−3 → H2O + CO2 k3 = 7 × 104 l mol−1 s−1 | (3) |
The detection and quantification limits could be improved using the same apparatus by increasing the counting times, using 2 mL of bubbler solution, and potentially using half of the volume of NaOH solution in the same bubbler design. Increasing the counting times to 30 min would bring the noise level down to the equivalent of 1.5 dpm, thus decreasing the quantification limit by a factor of two. Hence the overall detection limit could be lowered to 12 Bq L−1 and the quantification limit to ≡90 Bq L−1; this was not tested. We further tested the procedure by precipitating the dissolved CO2 of the bubbler solution as CaCO3,7,8 which could lower the detection limit to <1 Bq L−1 (to be published separately).
5 Conclusion
Our test method allowed for carbon-14 determination in a difficult matrix consisting of highly tritiated heavy water, taken from reactor systems. Removal of tritium was quasi-quantitative, while recoveries of carbon-14 were ≡94%, based on several runs with process standards and standards in simulated solutions. The method was tested with five analysts, and showed no operator bias. Key items were the choice of desiccant and the design of the reaction vessel, which could easily be custom-made at a low cost.The detection limit is sufficiently low to analyze contaminated groundwaters down to levels near the Canadian drinking water guideline of 200 Bq L−1. The reactor water samples had 14C levels sufficiently higher than this, and reliable quantification was possible.
6 Acknowledgements
The authors thank J. Torok, R. Rao, J. Young, L. Jones, AECL, W. Watson, who was on term at AECL, and A. O’Connor, Cambrian College, Sudbury, ON, for various aspects of discussion and laboratory help with liquid scintillation techniques. The work was supported by COG (CANDU® Owners Group) Working Project Information and Release WPIRs 1524 and 2727. The authors thank M. Totland, J. Gulens (AECL) and R. Laporte (Hydro-Québec, Gentilly nuclear station) for reviewing the manuscript. The main author received the IUPAC/Gendron award, given by the Canadian Society for Chemistry to support the presentation of this work at SAC 99.7 References
- L. Z. Liepins and K. W. Thomas, Radiat. Waste Manage. Nucl. Fuel Cycle, 1978, 10, 357 Search PubMed.
- R. R. Rao, Tritium and Carbon-14 in CANDU Reactors: Comparison of Their Production and Release Rates with those from Other Types of Reactors, COG-97-463-I, Atomic Energy of Canada Limited, Chalk River, ON, 1997. Search PubMed.
- G. M. Milton and R. M. Brown, A Review of Analytical Techniques for the Determination of Carbon-14 in Environmental Samples, AECL-10803; COG-93-335, Atomic Energy of Canada Limited, Chalk River, ON, 1993. Search PubMed.
- , Canadian Drinking Water Guidelines: Radiological Characteristics, 6th edn. (Web Page), 1995, available at http://www.hc-sc.gc.ca/ehp/ehd/catalogue/bch_pubs/dwgsup_doc/radio-e.pdf. Search PubMed.
- D. L. Massart, B. G. M. Vandeginste, S. N. Deming, Y. Michotte and L. Kaufman, Chemometrics: a Textbook, Elsevier, Amsterdam, 1988. Search PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, J. Wiley and Sons, New York, 2nd edn., 1981. Search PubMed.
- K. Pfeiffer, D. Rank and M. Tschurlovits, Int. J. Appl. Radiat. Isot., 1981, 32, 665 CrossRef CAS.
- S. J. Kramer, G. M. Milton and W. L. Watson, Comparison of Counting Techniques for 14C, COG-98-252-I, Atomic Energy of Canada Limited, Chalk River, ON, 1998. Search PubMed.
Footnotes |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| ‡ © Copyright Government of Canada. |
| This journal is © The Royal Society of Chemistry 2000 |