Microdialysis sampling with on-line microbore HPLC for the determination of tirapazamine and its reduced metabolites in rats†
Kieran J. McLaughlin‡, Alexander A. Faibushevich§ and Craig E. Lunte*
Department of Chemistry, University of Kansas, Lawrence, Kansas 66045, USA
First published on UnassignedUnassigned7th January 2000
Abstract
An on-line microdialysis microbore HPLC method is described for the determination of the bioreductive anti-tumor agent, tirapazamine (3-amino-1,2,4-benzotriazine-1,4-di-N-oxide, SR4233, WIN59075, Tirazone, TPZ) and its two major reduced metabolites, 3-amino-1,2,4-benzotriazine-1-N-oxide (SR4317) and 3-amino-1,2,4-benzotriazine (SR4330). Detection limits of 0.003 μM, 0.005 μM and 0.007 μM were obtained for tirapazamine, SR4317 and SR4330, respectively. Linear ranges of 0.011–20 μM, 0.017–20 μM and 0.025–20 μM for tirapazamine, SR4317 and SR4330 permitted quantitative analysis of all three compounds in microdialysis samples. Typical intra-day reproducibilities (n = 7) of 4.1% (tirapazamine), 6.6% (SR4317), 9.9% (SR4317), and 1.8% (tirapazamine), 2.4% (SR4317) and 2.6% (SR4330) were obtained at the 0.12 μM and 1.2 μM levels, respectively. Inter-day reproducibilities (n = 5) of 3.4% (tirapazamine), 1.8% (SR4317), 4.5% (SR4330) and 2.5% (tirapazamine), 2.5% (SR4317) and 1.7% (SR4330) were obtained at the 0.12 μM and 1.2 μM levels, respectively. The use of an on-line microdialysis HPLC system, permitted the determination of tirapazamine, SR4317 and SR4330 in blood and muscle tissue of rats with a high temporal resolution of sampling. The pharmacokinetics of tirapazamine and its metabolites were studied in the muscle and blood of rats previously administered an intraperitoneal dose of tirapazamine.
Introduction
The exploitation of intrinsic differences between normal and malignant tissues has been a driving force in the development of therapeutic agents for the treatment of cancer. Solid tumors are poorly perfused with blood with the result that they contain cells at lower levels of oxygenation than occurs in normal tissues. The presence of these regions of hypoxia have until recently represented a considerable obstacle to therapeutic regimens designed for the treatment of solid tumors. As a consequence, different strategies were developed to overcome the challenges presented by the hypoxic conditions. In recent years these same conditions have been used as a rationale for the development of therapeutic agents that are bioreductively activated and have a high selective toxicity towards solid tumors.3-Amino-1,2,4-benzotriazine-1,4-di-N-oxide (WIN59075, Tirazone, Tirapazamine, TPZ) is the lead compound of the benzotriazine di-N-oxide class of bioreductive antineoplastic agents.1 It has shown considerable hypoxic cell killing selectivity and is currently in Phase II and Phase III clinical trials.1,2 The highly selective toxicity of TPZ towards tumors is believed to derive from its ability to cleave cellular DNA.3 This cleavage is attributed to a radical species generated by enzymatic one electron reduction of the heterocycle. Recent evidence suggests that a hydroxyl radical is the major DNA-cleaving species generated by this reduction. In mammalian cells, TPZ is ultimately reduced to the two electron reduction product SR4317 (3-amino-1,2,4-benzotriazine-N-oxide) and the four electron reduction product, SR4330 (3-amino-1,2,4-benzotriazine), under anaerobic and to a lesser extent, under aerobic conditions.4
The determination of TPZ, SR4317 and SR4330 has been previously carried out using HPLC in conjunction with UV,5 diode array6 and dual-electrode amperometric detection7 schemes. Walton and Workman5 described the use of a reverse phase separation on a phenyl column for the determination of TPZ, SR4317 and SR4330 in methanolic extracts of plasma, urine and liver microsomal preparations. A limit of detection, defined as two times the signal-to-noise ratio, of 0.5–0.7 ng was achieved. Our laboratory has previously reported the use of dual-electrode amperometric detection in conjunction with microbore liquid chromatography for the determination of TPZ, SR4317 and SR4330 in microdialysis samples.7 As TPZ and its metabolites are all easily reduced at a carbon electrode, reductive amperometric detection provided high selectivity and low detection limits with the chromatographic analysis. Separation of the three compounds was achieved in 22 min by reverse phase chromatography on a C18 column. The detection limits, at three times the signal-to-noise ratio, were 70 nM, 50 nM and 50 nM for TPZ, SR4317 and SR4330, respectively. The method was applied as an off-line system to the analysis of microdialysis samples from blood, muscle and tumor tissue.4,7 Capillary electrophoresis (CE) in conjunction with laser-induced fluorescence (LIF) has been reported for the determination of TPZ and SR4317 in microdialysis samples.8 The use of CE-LIF8 permitted the determination of TPZ and its main reduced metabolite, SR4317, in under 60 s. The second reduction product, SR4330, was not detectable using this method. The method was used as part of an on-line microdialysis CE system for the determination of TPZ and SR4317 in rats.
In recent years, microdialysis sampling has been shown to be a powerful technique for the study of the pharmacokinetics and drug metabolism of a wide range of compounds in blood, muscle, tumor and other tissues.9–12 It is typically performed by implanting a semi-permeable dialysis membrane at the site of interest after which a sampling solution is perfused through the implanted membrane. Only low molecular weight species in the extracellular fluid can cross the dialysis membrane and be collected, thus eliminating any proteins from the sample matrix. This sample clean up has several important consequences for pharmacokinetics and drug metabolism studies involving microdialysis sampling. Firstly, as proteins are excluded from the dialysate, only the unbound fraction of a drug is sampled. Microdialysis sampling therefore permits the direct determination of the unbound, therapeutically important fraction of the drug. Secondly, the elimination of proteins from the sample effectively stops any further metabolism. Finally, the sample can be injected directly on to the HPLC column without any further sample pretreatment. As a result, microdialysis sampling can be coupled to on-line HPLC analysis. In an on-line microdialysis HPLC system for the study of pharmacokinetics, the dialysis perfusion rate, sample volume, chromatographic analysis time and required temporal resolution of the experiment are all interdependent. In previous studies of the pharmacokinetics and metabolism of TPZ using microdialysis sampling, samples were collected every 10 min and analyzed off-line.7 As the current HPLC method has an analysis time of 23 min, any on-line system incorporating it would have a minimum temporal resolution of 23 min. Such a temporal resolution is not sufficient for the determination of TPZ pharmacokinetics. Use of CE-LIF as part of an on-line microdialysis CE system has been shown8 to permit excellent temporal resolution. However, as SR4330 cannot be determined by this method, it too is not generally suitable. In this paper, the development of an on-line microdialysis microbore HPLC method providing short chromatographic analysis time and low detection limits is described for the determination of TPZ, SR4317 and SR4330. The system was used for the determination of TPZ and its metabolites in dialysates of muscle and blood tissue of rats.
Experimental
Reagents
The structures of the benzotriazines, TPZ, SR4317 and SR4330 are shown in Fig. 1. 3-Amino-1,2,4-benzotriazine-1,4-di-N-oxide (tirapazamine, TPZ), 3-amino-1,2,4-benzotriazine-1-N-oxide (SR4317), and 3-amino-1,2,4-benzotriazine (SR4330) were gifts from the Sterling Drug Company (New York, NY, USA). HPLC grade acetonitrile and methanol were obtained from Fisher Scientific (Fairlawn, NJ, USA). All other reagents were obtained from Sigma Chemical Co. (St. Louis, MO, USA). All chemicals used were analytical grade and used as received. All buffer solutions were prepared with water obtained from a Barnstead Nanopure water purification system (Boston, MA, USA). | ||
| Fig. 1 Structures of TPZ, SR4317 and SR4330. | ||
Chromatographic system
All gradient and isocratic HPLC analyses were performed using a reverse phase microbore system with spectrophotometric detection. The HPLC system consisted of two LC-6A pumps (Shimadzu Corp., Kyoto, Japan) controlled by an SCL-6A system controller (Shimadzu) and an SPD-6AV UV–VIS spectrophotometric detector (Shimadzu) with an 8 μl volume detection cell. For off-line injections, a Rheodyne (Cotati, CA, USA) 7125 injection valve with a 20 μl sample loop was used. When used in the on-line microdialysis–HPLC mode, all injections were made via a CMA/160 (Carnegie Medecin, NJ, USA) on-line injector with a 20 μl sample loop. Data collection was by a PE Nelson 900 series interface (PE Nelson Division, Perkin Elmer Corp., Norwalk, CT, USA) connected to a personal computer running PE Nelson Turbochrom Version 4.1.2. software. Separation of TPZ, SR4317 and SR4330 was achieved on a 5 μm C18 microbore column, 1 × 150 mm, (Metachem Technologies Inc., Torrance, CA, USA). The column was protected with a 5 μm ODS guard column, 1 × 14 mm, (Bioanalytical Systems, Inc., West Lafayette, IN, USA).The mobile phase for isocratic separation consisted of acetonitrile and 50 mM phosphate buffer, pH 2.8, in various combinations as described in the text. The buffer component of the mobile phase was filtered through a 0.22 μm MAGNA-R nylon filter (MSI, Westboro, MA, USA), mixed with acetonitrile and finally sonicated for 10 min prior to use. A flow rate of 90 μl min−1 was used for all isocratic chromatography.
In the gradient separation, mobile phase A consisted of 50 mM phosphate buffer, pH 2.8, and mobile phase B consisted of 15 + 85 (v/v) acetonitrile–50 mM phosphate buffer, pH 2.8. A linear gradient from 20% B to 50% B was run over a 7 min period, maintained for 5 min, after which the mobile phase was returned to 20% B over 5 min and finally allowed to equilibrate for 5 min. When operated in the gradient mode, the HPLC system was modified for use with a microbore column. Mixing of mobile phases A and B was achieved at 2 ml min−1 using a 20 μl internal volume PEEK mixing T (Upchurch Scientific, Apple Valley, MN, USA) after which the flow was split using a flow splitter. All gradient chromatography was performed at a total flow rate of 90 μl min−1.
Preparation of stock and standard solutions
Individual stock solutions of TPZ, SR4317 and SR4330 were prepared by dissolving known weights of the compounds in 100 ml of methanol and storing at −20 °C. Standard solutions of each compound were prepared on a daily basis by serial dilution from the stock solution into Ringer’s solution. All standard solutions were stored at 4 °C until use.A 1 mg ml−1 solution of TPZ was used for administration to animals as part of pharmacokinetics studies. This solution was prepared by dissolving 20 mg of TPZ in Ringer’s solution and stored at −20 °C until use. Prior to a pharmacokinetics study, the solution was thawed and then placed in an ultrasonic bath for 10–15 min in order to ensure complete dissolution of TPZ.
Method validation
System reproducibility was evaluated by analysis of two standard mixtures of the benzotriazines. The first mixture consisted of TPZ, SR4317 and SR4330 at 0.12 μM while the second mixture was at 1.2 μM. Intra-day and inter-day reproducibilities were determined by making seven injections of each standard mixture each day over a five day period.The limits of detection (LOD) and quantitation (LOQ) were determined at 3sB and 10 sB, where sB is the standard deviation of the background noise. The value of sB was evaluated from the magnitude of noise in an injection of blank blood dialysate over a representative section of the baseline where the analyte peak was expected, and covering 20 times the width at half height of the analyte peak.
Microdialysis sampling
Microdialysis sampling was performed using a CMA/100 micro-injection pump (Bioanalytical Systems) coupled to a microdialysis probe. The probe fabrication procedure has been previously described.13 The perfusion fluid was a Ringer’s solution consisting of 155 mM NaCl, 5.5 mM KCl and 2.3 mM CaCl2. A perfusion flow rate of 1 μl min−1 was used for all microdialysis studies.Calibration of microdialysis probes
All microdialysis probes were characterized in vitro by both recovery and delivery experiments before implantation. Characterizations were performed using a 0.25 μM mixture of TPZ, SR4317 and SR4330 in Ringer’s solution. In vitro extraction efficiencies were determined by recovery experiments under stirred conditions with the standard mixture maintained at 37 °C in a container in a constant temperature dry bath. The microdialysis probe was placed in the standard solution and perfused with Ringer’s solution at 1 μl min−1. In vitro delivery experiments were then performed under the same conditions except that Ringer’s solution was maintained at 37 °C under stirred conditions and the standard mixture was delivered through the probe. Only probes for which the extraction efficiency by recovery and delivery was not statistically different at the 95% confidence level were used for subsequent implantation.Implanted probes were calibrated in vivo by delivery experiment. In vivo delivery experiments were performed by implanting the probe in the tissue of interest, allowing the animal to recover from anesthesia and perfusing the probe with Ringer’s solution for at least 12 h. After this the probe was perfused at 1 μl min−1 with the 0.25 μM standard mixture of TPZ, SR4317 and SR4330 in Ringer’s solution. The probe extraction efficiency (EE) was then calculated as
 | (1) |
Surgical procedures
Male Sprague–Dawley rats (250–400 g) were supplied from the University of Kansas Animal Care Unit breeding colony. Housing and care of the animals prior to experimental use was provided by the Animal Care Unit. Housing and care of the animals during experimental procedures were performed according to Animal Use Statements approved by the University of Kansas Institutional Animal Care and Use Committee. Strict aseptic techniques were used throughout all surgical procedures. During all surgical procedures, rats were pre-anesthetized with 1% isoflurane and subsequently anesthetized with a mixture of ketamine–xylazine (100 mg + 10 mg per kg body weight). Body temperature was maintained by placing a heating table under the rat during surgery.Implantation of a linear PAN (polyacrylonitrile) probe into hind leg muscle was carried out by first shaving the hair and cleaning the skin on the back of the neck and the area over the hind leg muscle. A 4 cm long incision was made in the hind leg skin to expose the muscle for implantation of the probe. In addition, an incision was made in the shaved and cleaned skin at the back of the neck. The skin was carefully loosened from the underlying muscle along the edges of the incision to form a subcutaneous pocket for positioning of the inlet and outlet tubing of the probe. Using a surgical introducer, a tunnel was then created from the leg incision, over the shoulder and up to the neck incision. A 1 cm linear PAN microdialysis probe was implanted in the hind leg muscle through a 3 cm long 23 gauge needle. The inlet and outlet tubing were secured at the muscle surface with a drop of tissue glue and externalized at the back of the neck. The skin incisions were then closed using glue and stainless steel clips.
Implantation of a microdialysis probe in the bloodstream was carried out by exposing the jugular vein and making a small V-shaped incision in it. A 1 cm flexible Cuprophan probe was inserted via the incision and threaded through the jugular vein to the superior vena cava in a region close to the heart. The jugular vein was then ligated and the inlet and outlet tubing threaded under the skin and out the incision at the back of the neck.
After probe implantation the animal was put in a collar tethered to a liquid swivel suspended over the center of an awake animal containment system. At least 12 h were allowed for the animal to recover from surgery before an experiment was begun. During this time the probe was perfused with Ringer’s solution. The animal had access to food and water ad libidum throughout the study. A 2 mg kg−1 dose of TPZ (1 mg ml−1 in Ringer’s solution) was administered as an intraperitoneal (ip) bolus injection.
Results and discussion
The chromatographic conditions employed in the present study were initially based on those used for the dual amperometric method previously reported by our laboratory.7 The choice of the mobile phase composition for the dual amperometric detection method was dictated to a large extent by the need to resolve the TPZ peak from the early eluting endogenous material in the microdialysis sample. As a consequence, the organic composition of the mobile phase was limited to 2–3%, resulting in retention times of 21 and 23 min for SR4330 and SR4317, respectively.In the design of an on-line microdialysis–HPLC system offering high temporal resolution, the chromatographic analysis time should be as short as possible. To achieve this in the analysis of TPZ and its metabolites, it was necessary to reduce the retention time of SR4330 and SR4317 whilst maintaining adequate resolution of TPZ from the endogenous peak. The use of spectrophotometric detection was investigated with a view to achieving this.
Spectrophotometric detection
The UV–VIS spectra for TPZ, SR4317 and SR4330 are shown in Fig. 2. It can be seen that significant absorbance occurs for all three compounds in the region of 220–270 nm and to a lesser extent in the region of 380–460 nm. UV detection at 254 nm and visible detection at 420 nm were investigated to determine their potential use for the HPLC determination of TPZ, SR4317 and SR4330.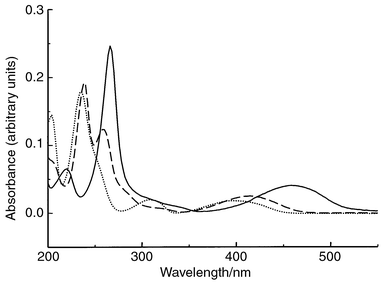 | ||
| Fig. 2 UV–VIS spectra of TPZ (—), SR4317 (---) and SR4330 (⋯). | ||
The use of UV detection at 254 nm permitted excellent sensitivity of analysis for standard mixtures of TPZ, SR4317 and SR4330. However, when applied to the analysis of dialysis samples obtained from rats not previously administered TPZ, numerous peaks originating from endogenous materials appear in the chromatogram, as shown in Fig. 3A. However, these chromatographic peaks were eliminated with the use of a detection wavelength of 420 nm as shown in Fig. 3B. The use of visible detection at 420 nm provided excellent limits of detection for standard mixtures of the three compounds, albeit less than was achieved with UV detection. More importantly however, with the use of visible detection at 420 nm, the large early eluting peaks characteristic of the electrochemical and UV detection schemes were completely removed from the chromatograms. The elimination of these background peaks removed the constraints imposed on mobile phase composition by the need to ensure their resolution from TPZ. The use of mobile phases with higher organic contents was now possible.
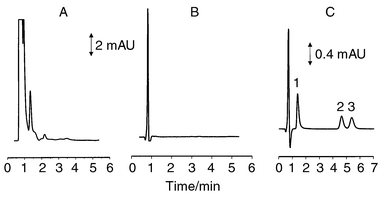 | ||
| Fig. 3 Chromatograms of blank blood dialysate at a detection wavelength of (A) 254 nm and (B) 420 nm. (C) Chromatogram of blank blood dialysate spiked with (1) TPZ, (2) SR4317 and (3) SR4330. Chromatographic conditions: 10 + 90 (v/v) acetonitrile–50 mM phosphate buffer, pH 2.8; flow rate 90 μL min−1. | ||
Mobile phase composition
With the emphasis on speed of analysis, the organic content of the mobile phase was increased incrementally from 3% to 15%. Increase in the organic composition from 3% to 10% allowed the time of analysis to be progressively decreased while still maintaining resolution of TPZ from the void peak and separation of SR4317 and SR4330. However, further increase of the organic content caused TPZ to elute in the void and incomplete separation of SR4317 and SR4330. A mobile phase containing 10% acetonitrile provided an analysis time of 6 min as shown in Fig. 3C. This mobile phase was therefore chosen for initial investigations on the fate and distribution of TPZ and its reduced metabolites, SR4317 and SR4330 in blood and muscle of rats. In Fig. 4, chromatograms are shown of blood dialysates obtained before and 90 min after ip administration of TPZ to a rat. These show excellent chromatography for TPZ and its two reduced metabolites with no obvious chromatographic interferences. However, in order to ascertain peak purity for each of the chromatographic peaks, gradient chromatography was carried out on blood dialysate samples collected before and after administration of TPZ to the animal. | ||
| Fig. 4 Chromatogram of (A) blank blood dialysate (B) dialysate obtained 90 min after ip administration of TPZ. Chromatographic conditions as in Fig. 3. | ||
Gradient HPLC of microdialysates
Blood microdialysis samples collected from a rat administered a therapeutic dose of TPZ were analyzed under the gradient chromatographic conditions. Chromatograms are shown in Fig. 5 for blood dialysates collected before and after an ip administration of TPZ. No additional peaks were observed in the blood dialysis sample collected before administration of TPZ. However, gradient chromatography of the blood dialysis sample collected after administration of TPZ revealed the presence of additional peaks eluting immediately after TPZ and before SR4330. As these peaks do not appear in the blank, they are assumed to be metabolites of TPZ. Based upon this observation, the conditions of the isocratic separation were modified to achieve resolution of all the components while keeping the analysis time to a minimum. To meet these requirements, a mobile phase composed of 7 + 93 (v/v) acetonitrile–50 mM phosphate buffer, pH 2.8, was chosen. An on-line chromatogram using these conditions is shown in Fig. 6. Baseline resolution of all components was achieved in a total analysis time of 12 min. While this is longer than the 6 min analysis time shown in Fig. 4, it does nevertheless represent a significant improvement over the 23 min analysis time for the method using electrochemical detection currently used in our laboratory. Moreover, the method provides the separation of three additional peaks, which are present in only those dialysis samples collected after TPZ administration to the animal under study. In the previously reported liquid chromatographic and capillary electrophoretic methods for the determination of TPZ, SR4317 and SR4330,7–10 no reference has been made to the presence of such peaks. These peaks have been consistently present in all animals studied to date using this chromatographic method and it is therefore probable that they are related to TPZ. However, in terms of the immediate goal of this study to develop an on-line microdialysis HPLC system, the 12 min time of analysis does permit adequate temporal resolution of sampling for our current research. | ||
| Fig. 5 Gradient HPLC chromatograms of (A) standard mixture of (1) TPZ, (2) SR4317 and (3) SR4330; (B) blood dialysate collected 90 min after TPZ ip administration. Chromatographic conditions: mobile phase A, 50 mM phosphate buffer, pH 2.8; mobile phase B, 15 + 85 (v/v) acetonitrile–50 mM phosphate buffer, pH 2.8. A linear gradient from 20% B to 50% B was run over 7 min, maintained for 5 min, after which the mobile phase was returned to 20% B over 5 min; flow rate 90 μL min−1. | ||
 | ||
| Fig. 6 Chromatograms of (A) blank blood dialysate and (B) blood dialysate collected 90 min after ip administration of TPZ. Chromatographic conditions: 7 + 93 (v/v) acetonitrile–50 mM phosphate buffer, pH 2.8; flow rate 90 μL min−1. | ||
As the HPLC method employing dual-electrode amperometric detection currently in use in our laboratory is prone to interference from oxygen the mobile phase must be frequently purged with and maintained under an inert gas. In addition, the electrodes are subject to passivation with continuous usage and require cleaning on a regular basis to maintain method sensitivity and reproducibility. The developed method employing visible detection circumvents these problems and consequently represents a more rugged system for the analysis of TPZ and its metabolites.
Linear range and limits of detection and quantitation
The linear range and limits of detection and quantitation were evaluated using a range of standards prepared on a daily basis by serial dilution of stock solutions into the appropriate volume of Ringer’s solution. The detection limits, defined as three times the standard deviation of the blank signal (3sB), were 0.003 μM, 0.005 μM, and 0.007 μM for TPZ, SR4317 and SR4330, respectively. These detection limits compare very favorably with those of both the dual-electrode amperometric method, where the limits of detection (3sB) were 0.070 μM, 0.050 μM and 0.050 μM for TPZ, SR4317 and SR4330, respectively,10 and 2 μM for TPZ using UV detection at 254 nm.6 The limits of quantitation, defined as ten times the standard deviation of the blank signal (10sB), were 0.011 μM, 0.017 μM and 0.025 μM for TPZ, SR4317 and SR4330, respectively. The detector response was linear over the range of 0.011–20 μM, 0.017–20 μM, and 0.025–20 μM for TPZ, SR4317 and SR4330, respectively.These linear range, detection and quantitation limit parameters provided a suitable working range for the in vivo study of the pharmacokinetics of TPZ and its metabolites in muscle and blood using the on-line microdialysis microbore HPLC system. When comparing the detection limits of the method reported here and those of the previously reported dual-electrode amperometric method it is necessary to take the injection volumes of 14 μl and 3.5 μl, respectively, into account. When calculated as the amount of material injected, the detection limits were 7.5, 11.3, and 14.3 pg of TPZ, SR4317 and SR4330, respectively for the HPLC method employing visible detection at 420 nm. Detection limits of 43.6, 28.4, and 25.6 pg were achieved for TPZ, SR4317 and SR4330, respectively using dual amperometric detection. These results show that the mass limits of detection for both methods were quite similar, and were in fact slightly better in the case of the spectrophotometric method.
Intra- and inter-day reproducibilities
The average intra-day and inter-day variations over five days are presented in Tables 1 and 2. Typical intra-day reproducibilities (n = 7) of 4.1% (TPZ), 6.6% (SR4317), 9.9% (SR4317), and 1.8% (TPZ), 2.4% (SR4317) and 2.6% (SR4330) were obtained at the 0.12 μM and 1.2 μM levels, respectively. Inter-day reproducibilities (n = 5) of 3.4% (TPZ), 1.8% (SR4317), 4.5% (SR4330) and 2.5% (TPZ), 2.5% (SR4317) and 1.7% (SR4330) were obtained at the 0.12 μM and 1.2 μM levels, respectively.| 0.12 μM | 1.2 μM | |||||
|---|---|---|---|---|---|---|
| Day | TPZ | SR4317 | SR4330 | TPZ | SR4317 | SR4330 |
| 1 | 2.57 | 5.02 | 10.35 | 2.22 | 3.32 | 3.07 |
| 2 | 5.13 | 6.79 | 8.68 | 1.71 | 2.12 | 2.68 |
| 3 | 5.40 | 7.93 | 9.74 | 1.05 | 2.41 | 2.46 |
| 4 | 2.98 | 7.03 | 11.01 | 2.29 | 2.19 | 2.49 |
| 5 | 4.56 | 6.28 | 9.66 | 1.53 | 1.78 | 2.26 |
| Mean | 4.13 | 6.61 | 9.89 | 1.76 | 2.36 | 2.57 |
| 0.12 μM | 1.2 μM | |||||
|---|---|---|---|---|---|---|
| Day | TPZ | SR4317 | SR4330 | TPZ | SR4317 | SR4330 |
| 1 | 10507 | 16193 | 10130 | 108609 | 165062 | 101574 |
| 2 | 11287 | 15699 | 9691 | 115992 | 172448 | 104392 |
| 3 | 11412 | 16164 | 10257 | 114504 | 175271 | 105981 |
| 4 | 11379 | 16467 | 10919 | 113124 | 174924 | 105739 |
| 5 | 11300 | 15955 | 9950 | 114338 | 174911 | 105479 |
| RSD (%) | 3.38 | 1.78 | 4.51 | 2.49 | 2.50 | 1.73 |
On-line in vivo studies
Fig. 7 shows the typical output obtained from an on-line microdialysis experiment on an awake, freely moving rat. The outlet of the microdialysis probe was directly connected to the HPLC injection valve. The dialysate solution continuously filled the 20 μl sample loop of the injection valve. An injection into the chromatographic system was made every 14 min for 1 min. This allowed 13 min for the dialysate to fill the sample loop which results in under-filling the sample loop. Therefore the injection volume was 13 μl as determined by the dialysis perfusion rate and sampling interval. Microdialysis samples were collected and analyzed for at least 30 min prior to administration of TPZ. After ip administration to the animal, the distribution and elimination of TPZ, SR4317 and SR4330 was followed for over three days. A representative section of the overall data set is shown in the inset of Fig. 7. This shows that the continuous data stream from the on-line system consists of a series of chromatograms representing the microdialysis sample collected over that particular sampling interval. From the expanded section, it can be seen that TPZ and its two major metabolites, SR4317 and SR4330 can be resolved along with several unidentified metabolites of TPZ. | ||
| Fig. 7 On-line chromatograms from an animal administered an ip dose of TPZ from time of TPZ administration at 50 min. Chromatographic conditions as in Fig. 6 except that the on-line injector was used. | ||
It is important to point out that this on-line microdialysis HPLC study on the pharmacokinetics of TPZ was limited in its scope. However, the results on the distribution and elimination of TPZ and its principal reduced metabolites, SR4317 and SR4330, are in agreement with results obtained from comprehensive studies in our laboratories using the dual amperometric methodology.7 As previously stated, this electrochemical detection scheme was used in an off-line mode with discrete dialysis samples being collected every 10–15 min and frozen until subsequent analysis. The on-line approach described here requires significantly less operator attention and allows real time results to be obtained. This latter point is of importance in our research on the effects of ischaemia and reperfusion on the in vivo metabolism of TPZ. The initiation and termination of ischaemic events, by clamping of blood vessels, is guided by the concentrations of TPZ and its metabolites present in collected samples at that time point. The real time monitoring capability of the on-line method provides the necessary feedback information allowing these initiation and termination events to be carried out in an expeditious and precise manner.
Conclusions
The development of an on-line microdialysis microbore HPLC method has been described for the determination of the benzotriazine anti-tumor agent, TPZ and its reduced metabolites SR4317 and SR4330 in rats. The use of spectrophotometric detection at 420 nm gave a chromatographic background free from the endogenous interferences characteristic of the electrochemical and UV detection schemes. This permitted the development of a chromatographic method providing separation and baseline resolution of TPZ, SR4317 and SR4330 in addition to three additional peaks consistently present in all animals administered therapeutic doses of TPZ. When compared to a previously reported and currently used dual amperometric detection scheme, the present method offered excellent limits of detection and quantitation in addition to good intra- and inter-day reproducibilities. When applied as part of the on-line microdialysis HPLC system to an in vivo analysis, the complete distribution and elimination of TPZ, SR4317 and SR4330 could be followed in a single, awake, freely moving animal. The method required a minimum of operator attention and provided real time results on the distribution of TPZ and its metabolites. The current on-line system has replaced a previously developed HPLC method incorporating dual amperometric detection8 for TPZ analysis in our laboratory. In addition, it has been successfully transferred to two research projects on TPZ and has, to date, proven extremely reliable and rugged.Acknowledgements
Financial support for this work was provided by grant R01-GM44900 from the National Institutes of Health.References
- C. J. Koch, Cancer Res., 1993, 53, 3992 Search PubMed.
- M. J. Dorie and J. M. Brown, Cancer Chemother. Pharmacol., 1997, 39, 361 Search PubMed.
- J. M. Brown and A. J. Giaccia, Cancer Res., 1998, 58, 1416 Search PubMed.
- R. K. Palsmeier and C. E. Lunte, Life Sci., 1994, 55, 815 CrossRef CAS.
- M. I. Walton and P. Workman, J. Chromatogr., 1988, 430, 429 CrossRef CAS.
- H. Robin, Jr., S. Senan and P. Workman, Cancer Chemother. Pharmacol., 1995, 36, 266 Search PubMed.
- X. Liang, R. Palsmeir and C. E. Lunte, J. Pharm. Biomed. Anal., 1996, 1, 40.
- B. L. Hogan, S. M. Lunte, J. F. Stobaugh and C. E. Lunte, Anal. Chem., 1994, 66, 596 CrossRef CAS.
- D. O. Scott and C. E. Lunte, Pharm. Res., 1993, 19, 335 CrossRef.
- W. F. Elmquist and R. J. Sawchuk, Pharm. Res., 1997, 14, 267 CrossRef CAS.
- M. I. Davies and C. E. Lunte, Chem. Soc. Rev., 1997, 26, 215 RSC.
- D. K. Hansen, M. I. Davies, S. M. Lunte and C. E. Lunte, J. Pharm. Sci., 1999, 88, 14 CrossRef CAS.
- M. I. Davies and C. E. Lunte, Life Sci., 1996, 59, 1001 CrossRef CAS.
Footnotes |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| ‡ Current address: Astra Zeneca, Wilmington, DE 19850, USA. |
| § Current address: Oread Laboratories, Lawrence, KS 66047, USA. |
| This journal is © The Royal Society of Chemistry 2000 |