Determination of cyclamate in low-calorie foods by high-performance liquid chromatography with indirect visible photometry
Martin M. F. Choi*, Mei Ying Hsu and Siu Lan Wong
Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China.. E-mail: mfchoi@net1.hkbu.edu.hk
First published on UnassignedUnassigned7th January 2000
Abstract
A rapid and simple method using reversed-phase high-performance liquid chromatography combined with indirect visible photometry at 433 nm was developed to determine cyclamate in some food samples. Cyclamate was not detected in these chosen samples as its use is banned in Hong Kong. Cyclamate can easily be detected in spiked samples using a mobile phase consisting of 30 μmol dm−3 Methyl Red and 0.02 mol dm−3 phosphate buffer (pH 7.0)–methanol in a volume ratio of 3∶2. The column temperature was set at 23 °C. The detection limit was 0.14 mmol dm−3 and the relative standard deviation of the peak area response was 0.58% for a solution containing 5.0 mmol dm−3 of cyclamate (n = 8). This method was successfully applied to the analysis of eight spiked food samples and the cyclamate recoveries for these samples ranged from 93 to 99%.
Artificial sweeteners play an important role in our society not only for diabetic patients but also for people using low-calorie foods. To date the most commonly used artificial sweeteners are saccharin, cyclamate, aspartame and acesulfam. The sodium and calcium salts of cyclamic acid are used extensively in many diet and medical products. It has been reported that cyclamate is a potent carcinogen when it is converted into cyclohexylamine in the gastrointestinal tract. Some countries such as Canada, USA, European countries, India and Hong Kong have banned its use as a food additive but its usage is still permitted in other countries including mainland China.1,2 The detection and quantification of cyclamate in diet foods is still regularly performed in Hong Kong.
Tremendous efforts over the years have been devoted to the development of methods for the determination of cyclamate. There are many analytical procedures for determining this sweetener, such as gravimetry,3 volumetric analysis,4 amperometry,5 ion-selective electrode,6 gas chromatography,7 high-performance liquid chromatography (HPLC),8–12 capillary electrophoresis13 and spectrophotometry or flow injection spectrophotometry.14–17 Most of these methods require extensive or laborious chemical reactions and extraction procedures. HPLC is one of the most useful and popular techniques as it can separate target analytes from a complex mixture in samples when an optimum mobile phase and stationary phase are employed. Unfortunately, traditional HPLC is not suitable for the determination of cyclamate as cyclamate does not absorb in the usable ultraviolet/visible (UV/VIS) range. This shortcoming has been solved by using indirect UV photometry,8,13 post-column ion-pair extraction,9,10 pre-column derivatisation11 and conductivity detection.12 Indirect UV photometry has been shown to be a very sensitive detection method and has the advantage of being applicable to conventional HPLC instrumentation without any cumbersome modification.18 In this technique, a fixed concentration of a UV absorbing ion is added into the mobile phase. The absorbance of this mobile phase is then monitored and solute ions can easily be detected by the change in the baseline absorbance as either negative or positive peaks.19,20 In most indirect photometric methods, UV absorbing ions are normally used as they are compatible with the UV light sources such as deuterium, mercury arc and xenon lamps used in HPLC. However, the use of chromogenic dyes is seldom mentioned or realised. It is possible that the principle of indirect photometry in conjunction with HPLC can also be applied to a mobile phase containing a visible absorbing ion. The main advantage of using visible light detection is that the latest optoelectronics including super-bright light emitting diodes (LEDs), photodiodes, charge-coupled devices (CCDs) and plastic optical fibres can be used to construct a relatively cheap and compact detection unit for HPLC.
In this paper, we demonstrate the use of a chromogenic dye, o-Methyl Red, as a visible absorbing dye dissolved in a mobile phase, combined with an octadecylsilica column to determine cyclamate in various low-calorie foods.
Experimental
Materials
Aspartame, cyclamate sodium salt and saccharin sodium salt were obtained from Sigma (St. Louis, MO, USA), o-Methyl Red sodium salt from Aldrich (Milwaukee, WI, USA), methanol (HPLC grade) from Acros Organics (Geel, Belgium) and disodium hydrogen phosphate and monosodium dihydrogen phosphate from Farco Chemical Supplies (Beijing, China). All other reagents were of analytical-reagent grade and mobile phase solutions were prepared in de-ionised water.Instrumentation
The high-performance liquid chromatograph used was an HP1050 series consisting of a pumping system, a vacuum degasser and a variable wavelength detector (Hewlett-Packard, Wilmington, DE, USA). The column was an Inertsil ODS-3, 5 μm particle size, 250 × 4.6 mm id (GL Sciences, Tokyo, Japan). The column temperature was controlled by a Model CH-30 column heater in conjunction with a Model TC-50 temperature controller (Eppendorf Scientific, Westbury, NY, USA). pH measurements were taken on an Orion (Chicago, IL, USA) combined pH glass electrode. UV/VIS absorption spectra were measured on a Cary 100 Scan UV/VIS spectrophotometer (Varian Australia, Mulgrave, Victoria, Australia) using 10 mm matched cuvettes.Chromatographic procedures
Mobile phases were prepared from solvent mixtures of methanol and solutions of Methyl Red dissolved in phosphate buffers (pH 7.0). The mobile phase compositions were varied with different volume ratios of methanol and phosphate buffers and also concentrations of Methyl Red in the buffers. All mobile phases were filtered through a 0.45 μm filter and degassed in an ultrasonic bath prior to use. Normally, the column was pre-conditioned for 1–2 h by pumping through a mobile phase at a flow rate of 1.0 cm3 min−1 and the column effluent was monitored at 433 nm. The chromatographic separation was then begun after a steady baseline response of the detector had been attained. The calibration standards were dissolved in the mobile phases and injected into a Rheodyne sample injector having a sample loop volume of 20 mm3. Spiked samples were prepared by either dissolving in or diluting with the mobile phases, if not stated otherwise.Results and discussion
Selection of wavelength for detection
o-Methyl Red is a well known chromogenic dye which has a strong UV/VIS absorption spectrum. The UV/VIS absorption spectrum of a chromogenic dye may shift on changing the solvent system used. Therefore, the UV/VIS absorption spectrum of Methyl Red in mobile phases of various compositions were determined and are displayed in Fig. 1. The UV/VIS absorption spectrum does not shift much when the volume ratio of buffer to methanol is varied from 4∶1 to 1∶1. The absorption peak maximum is at 433 nm and was used throughout the chromatographic separation.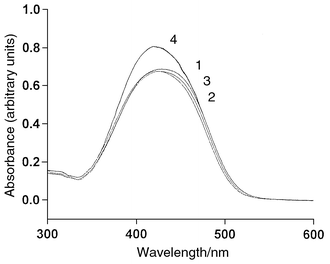 | ||
| Fig. 1 Absorption spectra of Methyl Red (30 μmol dm−3) in various volume ratios of 0.02 mol dm−3 phosphate buffer (pH 7.0)–methanol: (1) 4∶1; (2) 7:3; (3) 3∶2; (4) 1∶1. | ||
Selection of eluent pH
The use of o-Methyl Red as an indicator for acid–base titrations in aqueous solutions is based on the observation that the indicator changes its colour in the pH range 4.2–6.3.21 The acid dissociation constant of o-Methyl Red is pKa = 5.1.22 It is obvious that different forms of Methyl Red exist in different pH ranges. In Fig. 1, broad absorption bands at 350–500 nm are observed due to the formation of the yellow anionic form of Methyl Red at pH 7.0. This dominant anionic form does not change even on addition of methanol to the buffer and is consistent with the results obtained by other researchers.23 An intense absorption band with a peak maximum of 520 nm is observed if the pH is lowered to 3.8 because of the formation of the red protonated form of Methyl Red.24 In principle, the red protonated form of Methyl Red can also be used as the visible-absorbing ion in the mobile phase for indirect photometric detection.25 Unfortunately, the light intensity of our HPLC detection system at 520 nm is not strong enough for photometric measurement. Therefore, the pH of the eluent was kept at 7.0 in order to ensure that all the Methyl Red was in its anionic form. pH values higher than 7.0 are not recommended as there is a possibility of the dissolution of silica material in the column at high pH values.Selection of buffer and its concentration
The use of buffer is essential to keep the eluent pH constant as Methyl Red can exist in different forms at different pH values. There are many buffer systems that can be tried, such as sodium hydrogen carbonate–carbonic acid, ammonium chloride–aqueous ammonia and sodium phosphate buffers. However, the last one is the most appropriate choice because phosphate buffer has the advantage of reducing any other mixed mechanisms that lead to peak tailing. Phosphate salt can effectively compete for the electrostatic sites by hindering the ionic interactions between sample solutes and uncapped silanol groups of the packing materials.26 Second, a wide pH range from 4 to 12 can easily be prepared when a phosphate buffer is used.Different concentrations of phosphate buffers (0–0.2 mol dm−3) at pH 7.0 were used to prepare 3∶2 buffer–methanol. It was found that a lower concentration of buffer gave a very unstable baseline for detection. However, too high a concentration of buffer will build up a high column back-pressure, which can damage the operation of an HPLC pump. There is also a risk of precipitating the phosphate salt if a high concentration of buffer is used. From our experience, an optimum concentration of phosphate buffer of 0.02 mol dm−3 was chosen throughout the remainder of the experiments.
Composition of mobile phase
The volume ratio of a buffer–methanol mixture as the mobile phase was investigated for chromatographic separation of artificial sweeteners. When a volume ratio of 9∶1 of buffer to methanol was used as the mobile phase, there was no background absorption signal on the detector. Methyl Red was completely retained in the column since the mobile phase is too hydrophilic. However, when 100% methanol was pumped through the column again, all the Methyl Red was desorbed from the column, giving a very high background absorption signal. Lower volume ratios of buffer–methanol mixtures as mobile phases (4∶1 to 1∶1) were subsequently tested. It was found that at 4∶1 and 7∶3 volume ratios of buffer to methanol, the saccharin, cyclamate and aspartame peaks were not satisfactorily separated. When the volume ratio was set at 3∶2, all the solute peaks were completely separated within 12 min.Fig. 2 shows a typical chromatogram for the separation of a mixture of saccharin, cyclamate and aspartame using buffer–methanol (3∶2) as the mobile phase and o-Methyl Red as the visible-absorbing anion. The first negative peak was assigned as the injection peak arising from a decrease in absorbance because of all the unretained solutes and the eluent dilution resulting from the injected sample solvent. This observation was confirmed by the injection of a solvent without cyclamate into the column. A similar negative peak at the same retention time was found in the chromatogram. The second, third and fourth positive peaks were identified as saccharin, cyclamate and aspartame, respectively. The retention mechanisms for these sweeteners are complicated processes. It has been mentioned that dynamic modification of a hydrophobic surface of a reversed stationary phase to a charged double layer can be achieved by the sorption of a hydrophobic ionic modifier from the eluent on its hydrophobic surface.27 In the present mobile phase system, it is probable that Methyl Red with its counter-ion (Na+) is adsorbed on the octadecylsilica surface to form a charged double layer. Below pH 7.0, cyclamate and aspartame are present in anionic form whereas saccharin is in neutral form. The retention of cyclamate and aspartame can be explained by the anion exchange mechanism between the analyte and the surface adsorbed Methyl Red ions.28 However, the retention of saccharin is possibly based on the displacement of the sodium–Methyl Red ion-pair from the stationary phase.
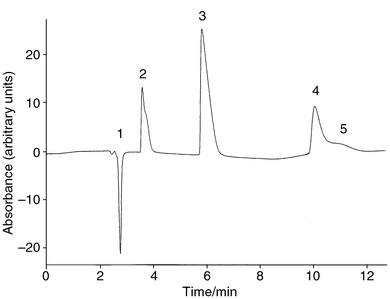 | ||
| Fig. 2 Chromatogram showing the separation of saccharin (4.9 mmol dm−3), cyclamate (5.0 mmol dm−3) and aspartame (3.4 mmol dm−3) using a mobile phase of 3∶2 buffer (pH 7.0)–methanol and 30 μmol dm−3 Methyl Red at a column temperature of 23 °C. (1) Injection peak; (2) saccharin; (3) cyclamate; (4) aspartame; (5) system peak. | ||
In general, the sample elution peaks can be either positive or negative according to their charge and retention relative to the UV/VIS-absorbing ion. The injection of a sample with the same charge as the visible-absorbing ion (in the present system) gives a positive peak if the capacity factor of the sample is smaller than the visible-absorbing ion.25 In addition to the sample solute peaks, a small positive system peak eluting close to the aspartame peak was also observed. This system peak is considered to arise from the elution of neutral eluent molecules desorbed from the column surface during sample injection. The occurrence of this system peak can be explained by reversed-phase and dye-displacement mechanisms.29 Apparently, the present eluent still contained a very small amount of neutral molecules. If the eluent pH is raised to a level where the eluent is completely ionised, then the system peak can be eliminated.20 However, the pH of the present mobile phase cannot be raised further as dissolution of the packing material occurs, as mentioned above.
When the composition of the mobile phase was changed to 1∶1 buffer–methanol, a standard mixture of saccharin, cyclamate and aspartame could also be completely separated within 7 min. However, the sensitivity for detection of cyclamate is lower than that when using a mobile phase of 3∶2 buffer–methanol. Hence the sample analysis of low-calorie foods was only carried out using 3∶2 buffer–methanol.
Effect of Methyl Red concentration
The effect of Methyl Red concentration on the sensitivity for the detection of cyclamate was investigated by varying its concentration in the mobile phase from 10 to 50 μmol dm−3. A series of cyclamate standards were injected into the column. The peak area was plotted against the concentration of cyclamate and the results for the regression analysis are given in Table 1. The higher the concentration of the Methyl Red in the mobile phase, the higher was the sensitivity for the detection of cyclamate. However, at a higher dye concentrations, it would generate a higher background absorption signal, which in turn would increase the noise level of the detection. It has been suggested that the photometric error would be smaller if the absorbance range can lie between 0.2 and 0.8.30 Therefore, the eluent ion concentration (10–30 μmol dm−3) chosen should have a background absorption that lies within this range. The limit of detection can be determined as three times the standard deviation of the peak area of the blank and from results for the linear regression equations in Table 1.31 The limits of detection for 10, 30 and 50 μmol dm−3 Methyl Red solvent systems were 0.80, 0.14 and 0.30 mmol dm−3, respectively. Consequently, a concentration of Methyl Red of 30 μmol dm−3 was chosen for most chromatographic separations so as to maximise the sensitivity of detection, minimise the photometric error and lower the limit of detection for cyclamate.| Methyl Red concentration/μmol dm−3 | Slope/area unit mmol−1 dm3 | y-Intercept/area unit | r2 |
|---|---|---|---|
| 10 | 39.33 ± 0.60 | −4.73 ± 5.19 | 0.9988 |
| 30 | 101.64 ± 0.28 | 8.32 ± 2.40 | 0.9999 |
| 50 | 153.57 ± 0.91 | 22.52 ± 7.94 | 0.9998 |
Effect of column temperature
It has been mentioned that careful temperature control of the whole chromatographic system is required for good stability of the mobile phase absorbance when indirect photometry is applied.25 The effect of column temperature from 23 to 32 °C on the sensitivity for the detection of cyclamate was therefore investigated. A series of cyclamate standards were injected into the HPLC system at different column temperatures. The peak area was then plotted against the concentration of cyclamate and the results for the regression analysis are given in Table 2. The sensitivity increases with increase in temperature up to 32 °C. The sensitivities at 27 and 32 °C are very close to each other. Although higher temperatures can improve the sensitivity slightly, it was found that the limit of detection was worse at higher column temperatures (0.14, 0.35 and 0.40 mmol dm−3 cyclamate at 23, 27 and 32 °C, respectively). Therefore, the column temperature was set at 23 °C for the determination of cyclamate in food samples.| Column temperature/°C | Slope/area unit mmol−1 dm3) | y-Intercept/ area unit | r2 |
|---|---|---|---|
| 23 | 101.64 ± 0.28 | 8.32 ± 2.40 | 0.9999 |
| 27 | 117.70 ± 0.79 | 9.05 ± 6.82 | 0.9998 |
| 32 | 115.92 ± 0.88 | 15.15 ± 7.62 | 0.9997 |
Repeatability
In order to investigate the repeatability of the proposed method, a 5.0 mmol dm−3 cyclamate standard was injected eight times into the HPLC system using optimum conditions of 30 μmol dm−3 Methyl Red, 3∶2 buffer–methanol and 23 °C. The relative standard deviation was found to be 0.28%. Our proposed HPLC method demonstrates good repeatability.Analysis of food samples
Eight commercial samples were bought from a local supermarket in Hong Kong and analysed using the optimum experimental conditions. No detectable cyclamate was found in these samples. Recovery tests were also used to test the validity of our method by adding a fixed amount of cyclamate to the food samples. A typical chromatogram of a spiked sample (Diet Seven-up) is shown in Fig. 3. A small positive peak that eluted before the cyclamate peak was identified as benzoate. This is not surprising since benzoic acid is a commonly used preservative added to soft drinks. A small amount of aspartame was also found in the chromatogram, as aspartame is added as a low-calorie sweetener in this type of sample. Chromatograms for other spiked samples were very similar. The recovery tests for all the spiked samples are summarised in Table 3 and the results were satisfactory.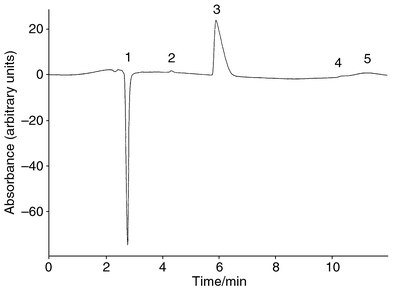 | ||
| Fig. 3 Chromatogram of Diet Seven-up spiked with 5.0 mmol dm−3 cyclamate using a mobile phase of 3∶2 buffer–methanol containing 30 μmol dm−3 Methyl Red at pH 7.0 and 23 °C. (1) Injection peak; (2) benzoate; (3) cyclamate; (4) aspartame; (5) system peak. | ||
Conclusion
An HPLC method combined with indirect visible photometry for the separation and determination of cyclamate was developed. The method was applied to the analysis of real samples with satisfactory results. There is great potential for employing indirect visible photometry in HPLC since the latest optoelectronics, including super-bright LEDs, CCDs, photodiodes and optical fibres, can easily be incorporated to assemble a compact and inexpensive detection system for HPLC instrumentation.Acknowledgements
The research described in this paper was made possible in part by the HKBU (project No. FRG/97-98/II-05).References
- Encyclopaedia of Food Science, Food Technology and Nutrition, ed. R. Macrae, R. K. Robinson and M. J. Sadler, Academic Press, London, 1993, vol. 2, p. 1287. Search PubMed.
- M. Mathlouthi and C. Bressan, in Low-Calorie Foods and Food Ingredients, ed. R. Khan, Blackie, Glasgow, 1993, ch. 8. Search PubMed.
- J. B. Wilson, J. Assoc. Off. Anal. Chem., 1955, 38, 559 Search PubMed.
- M. L. Richardson and P. E. Luton, Analyst, 1966, 91, 520 RSC.
- O. Fatibello-Filho, M. D. Capelato and S. A. Calafatti, Analyst, 1995, 120, 2407 RSC.
- D. Feng and C. Chen, Fenxi Huaxue, 1988, 16, 737 Search PubMed.
- J. A. W. Dalziel, R. M. Johnson and A. J. Shenton, Analyst, 1972, 97, 719 RSC.
- A. Herrmann, E. Damawandi and M. Wagmann, J. Chromatogr., 1983, 280, 85 CrossRef CAS.
- J. F. Lawrence, Analyst, 1987, 112, 879 RSC.
- J. F. Lawrence and C. F. Charbonneau, J. Assoc. Off. Anal. Chem., 1988, 71, 934 Search PubMed.
- I. Casals, M. Reixach, J. Amat, M. Fuentes and L. Serra-Majem, J. Chromatogr. A, 1996, 750, 397 CrossRef CAS.
- Q.-C. Chen, S.-F. Mou, K.-N. Liu, Z.-Y. Yang and Z.-M. Ni, J. Chromatogr. A, 1997, 771, 135 CrossRef CAS.
- C. O. Thompson, V. C. Trenerry and B. Kemmery, J. Chromatogr. A, 1995, 704, 203 CrossRef CAS.
- K. C. Güven, T. Özol, N. Ekiz and T. Güneri, Analyst, 1984, 109, 969 RSC.
- S. T. Gouveia, O. Fatibello-Filho and J. de A. Nóbrega, Analyst, 1995, 120, 2009 RSC.
- C. S. P. Sastry, K. R. Srinivas, K. M. M. K. Prasad and A. G. Krishnamacharyulu, Analyst, 1995, 120, 1793 RSC.
- C. Cabero, J. Saurina and S. Hernández-Cassou, Anal. Chim. Acta, 1999, 381, 307 CrossRef CAS.
- H. Small and T. E. Miller, Jr., Anal. Chem., 1982, 54, 462 CAS.
- M. Denkert, L. Hackzell, G. Schill and E. Sjsgren, J. Chromatogr., 1981, 218, 31 CrossRef CAS.
- P. E. Jackson and P. R. Haddad, J. Chromatogr., 1985, 346, 125 CrossRef CAS.
- A. I. Vogel, Vogel’s Text-book of Quantitative Inorganic Analysis Including Elementary Instrumental Analysis, Longman, London, 4th edn., 1978, p. 761. Search PubMed.
- D. F. Boltz and J. A. Howell, Colorimetric Determination of Nonmetals, John Wiley and Sons, New York, 2nd edn., 1978, p. 60. Search PubMed.
- S. A. Tucker, H. C. Bates and W. E. Acree, Jr., Analyst, 1995, 120, 2277 RSC.
- K. M. Tawarah and H. M. Abu-Shamleh, Dye Pigm., 1991, 17, 203 Search PubMed.
- CRC Handbook of HPLC for the Separation of Amino Acids, Peptides, and Proteins, ed. W. S. Hancock, CRC Press, Boca Raton, FL, 1984, vol. 1, pp. 182–183. Search PubMed.
- B. A. Bidlingmeyer, Practical HPLC Methodology and Applications, John Wiley and Sons, New York, 1992, p. 147. Search PubMed.
- R. M. Cassidy and S. Elchuk, J. Chromatogr. Sci., 1983, 21, 454 CAS.
- B. B. Wheals, J. Chromatogr., 1987, 402, 115 CrossRef CAS.
- N. Chauret and J. Hubert, J. Chromatogr., 1989, 469, 329 CrossRef CAS.
- J. Weiss, Ion Chromatography, VCH, New York, 2nd edn., 1995, pp. 316–317. Search PubMed.
- J. C. Miller and J. N. Miller, Statistics for Analytical Chemistry, Ellis Horwood, Chichester, 3rd edn., 1993, pp. 115–118. Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |