Direct detection of large fat-soluble biomolecules in solution using membrane inlet mass spectrometry and desorption chemical ionization
Frants R. Lauritsen*, Maria Anita Mendes† and Tenna Aggerholm
Physical Biochemistry Group, Institute of Biochemistry, University of Southern Denmark—Odense University, DK-5230, Odense M, Denmark.. E-mail: frants@dou.dk
First published on UnassignedUnassigned7th January 2000
Abstract
This paper presents the first membrane inlet mass spectrometry system capable of detecting large biomolecules, such as testosterone (Mr 288), testosterone acetate (Mr 330) and α-tocopherol (Mr 430, vitamin E). The result was obtained using a home-made chemical ionization ion source with a thermostated tubular silicone membrane mounted right in the centre of a methane CI plasma. The liquid sample was flushed through the inside of the membrane for a period of 20–25 min, where the analyte diffused into the membrane. Following this trapping period the analyte was released from the membrane into the mass spectrometer by the combined action of heat radiation from the filament and charge transfer from the chemical ionization plasma. As a result of this stimulated desorption a good desorption peak was obtained as the analyte vaporized out of the membrane. Retinol (Mr 286, vitamin A), cholecalciferol (Mr 384, vitamin D3) and cholesterol (Mr 386) were also detected. However, these compounds (all containing a long hydrocarbon chain and being aliphatic alcohols) did not give a protonated molecule. They gave a series of cluster ions with the dominant located 20 mass units below the molecular ion. The detection limits of the new desorption chemical ionization MIMS technique were at low or sub-micromolar concentrations (high ppb levels) and the reproducibility was within 20%, when the area of the desorption peak was used for quantitation.
Introduction
Membrane inlet mass spectrometry (MIMS) has become an established technique for the monitoring1,2 and characterization3 of volatile metabolites in biological systems. The major advantage of the technique is the extreme ease with which samples can be analysed, whereas the disadvantage is the limitation to the detection of volatile and non-polar compounds. The easy sample handling (practically no pretreatment at all) has inspired several research groups to work on expansion of the technique to include larger and less volatile compounds. Most of this work has been reviewed recently.4,5The hitherto most successful method when it comes to the detection of semi-volatile (bp >250 °C) biological molecules is the trap-and-release method developed by Lauritsen’s group.6,7 In this method the sample liquid is flushed through a tubular silicone membrane mounted in the centre of the ion source of the mass spectrometer. Semi-volatile compounds diffuse into the membrane but do not evaporate significantly into the ion source before the membrane is rapidly heated (25–300 °C in 45 s) by radiation from the filament. In this fashion a useful desorption peak can be monitored. With the trap-and-release method relatively polar compounds such as phenoxyacetic acid (bp 285 °C), pentachlorophenol (bp 310 °C), acetylsalicylic acid (mp 135 °C) and caffeine (mp 238 °C) can be detected at low or sub μM concentrations.
MIMS has also found widespread use for the analysis of environmental samples8 and here researchers have developed methods capable of detecting the environmentally important polyaromatic hydrocarbons (PAHs). These compounds have extremely high boiling points (300–600 °C), but the hydrophobic nature of PAHs makes them very soluble in a silicone membrane and very low detection limits (≡1 nM) can be obtained. Like the trap-and-release method the methods for PAH detection are also based upon a stimulated desorption of compounds from the membrane surface. One method, developed by the group of Matz,9 is basically similar to the trap-and-release method except that the membrane is mounted outside the ion source and uses a traditional heating system. The latest and probably most promising method developed by Manish et al.10,11 uses lasers to stimulate the vaporization of non-volatile PAHs from the membrane surface and to ionize them selectively.
One of the major problems in connection with the detection of biomolecules using MIMS is the high polarity of many of them. Polar compounds do not dissolve very well in the silicone membrane and alternative membranes have to be used. However, it is not straightforward to replace the silicone membrane with a hydrophilic membrane, since as the membrane becomes more polar, the relative amount of water entering the system increases drastically and vacuum considerations become limiting. In addition the advantage of the enrichment of analyte as compared with water disappears. To overcome these problems microporous membranes were introduced by Lauritsen and Cooks.12 The microporous membranes only have limited selectivity but give a much higher flow of vapour into the instrument. In fact, the flow becomes sufficiently high for the vaporized water to be used as the reaction gas in CI. With this method small polar molecules like lactic acid and butane-2,3-diol can be monitored at nM concentrations. The solvent CI system was recently improved by Ketola and Lauritsen,13 who mounted a polyacrylonitrile membrane in the centre of the CI plasma and combined it with trap-and-release in a system similar to the desorption chemical ionization method developed by Baldwin and McLafferty.14 In this fashion they succeeded in the detection of the highly polar dicarboxylic acids malonic acid and succinic acid.
It is the results obtained with desorption chemical ionization (DCI-MIMS) right from the membrane surface that have led to the work presented here. In the first version of the system, described above, the water vapour passing through the membrane was used as the reaction gas for CI, but as has been described12 the high water pressure is detrimental to the filament. Therefore, we rebuilt the ion source in a fashion that allows a CI gas to be introduced into the ion source via a leak valve and equipped the membrane inlet with a thermostating unit. To test the system we replaced the polyacrylonitrile membrane with the traditional silicone membrane and hoped that the desorption chemical ionization idea would make detection of large, fat soluble compounds like cholesterol, the fat soluble vitamins and some cholesterol derived hormones possible. The ultimate goal of this research was a membrane inlet system capable of a fast and direct screening of blood samples for the presence of steroid hormones, etc.
Experimental
The DCI-MIMS experiment
The DCI-MIMS system developed for this work is similar to an earlier DCI-MIMS system described in detail elsewhere,13 except that a chemical ionization gas inlet line was attached to the ion source, and a thermostating unit was mounted on the sample inlet line at the entrance to the ion source (see Fig. 1). The separate CI gas inlet line allowed us to create a stable and well-defined CI plasma (methane) right on the membrane surface. In this way we were able to use a silicone rubber membrane (wall thickness 214 μm, Technical Products Inc., Decatur, GA, USA) and take advantage of the favourable participitation of the fat soluble biomolecules in the membrane as compared with the sample water. The thermostating unit, which was operated at a temperature a little higher than 100 °C, ensured that the large biomolecules could diffuse into the membrane at an acceptable rate during the sampling period. | ||
| Fig. 1 Photograph of the DCI-MIMS ion source. 1, Membrane; 2, filament; 3, ion source; 4, CI gas inlet; 5, thermostating of the sample liquid; 6, sample flow in; and 7, sample flow out. | ||
A typical experiment can be described as follows. The sample liquid is sucked through the membrane inlet at a rate of 1 ml min−1 for 20–25 min. During this period the large biomolecules diffuse into the membrane, but do not evaporate significantly into the ion source. After the sampling period a 3-way valve is used to switch from the sample liquid to atmospheric air, which is then passed through the membrane inlet for 90 s. When the liquid inside the membrane is replaced by air the membrane starts to heat as a result of heat radiation from the filament and a final temperature of about 300 °C is achieved. As a result of the combined action of increased temperature and charge transfer from the CI plasma, analyte molecules can be detected as they vaporize out of the membrane and create a desorption peak. The analyte concentration in the sample solution can then be determined as the area of the desorption peak. Following the desorption of analyte from the membrane, pure water is flushed through the inlet in order to cool the membrane down again. As long as the desorption period was kept shorter than 2 min no deterioration of the membrane was observed.
Other conditions
The mass spectrometer was a Balzers (Hudson, NH, USA) QMG 400 single quadrupole instrument with a mass range of 1–500. The total pressure of the methane CI gas was 7 × 10 −4 Torr in the vacuum chamber surrounding the ion source and estimated to be 0.3 Torr inside the ion source. To make sure that the methane CI conditions were perfect, a CI spectrum of ethyl hexanoate was recorded every morning. This spectrum (see Fig. 2) was obtained in a standard membrane inlet experiment simply by flushing the sample solution (5 mg L−1) through the membrane inlet for a few minutes without the use of an air-plug to induce membrane heating. The CI spectrum shows no surprises with [M − C2H4]+ at m/z 117 as the dominant ion and an intense (60%) ion at m/z 145, the protonated molecule [M+H]+. For comparison, the EI spectrum of ethyl hexanoate has only low abundance ions (less than 5%) at m/z values higher than 110 Dalton.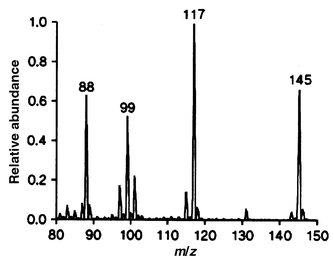 | ||
| Fig. 2 Methane CI mass spectrum of ethyl hexanoate obtained with the home-made chemical ionization ion source and standard membrane inlet technique. | ||
Chemicals were all commercially available from Sigma Chemical Company (St. Louis, MO, USA) and the samples were prepared by dilution into distilled water from 5 mM stock solutions in ethanol.
Results and discussion
Characterization of the DCI-MIMS spectra
Fig. 3(a) shows the desorption profile of testosterone (50 μM solution) recorded after a 25 min sampling period. The sample flow was replaced by the 90 s long air plug as indicated by the arrow. After 25 s delay (reflecting the transport of the air plug to the membrane) the signal increased rapidly for about 25 s before it slowly decreased again. Not all the analyte will have left the membrane at the termination of the air plug. The attachments of the membrane to the steel tubes are not sufficiently heated during the desorption process to induce the vaporization of analyte molecules trapped in this part of the membrane. Instead analyte molecules diffuse slowly from these parts towards the hot membrane centre where they heat up and vaporize. Memory effects in this connection will be discussed later.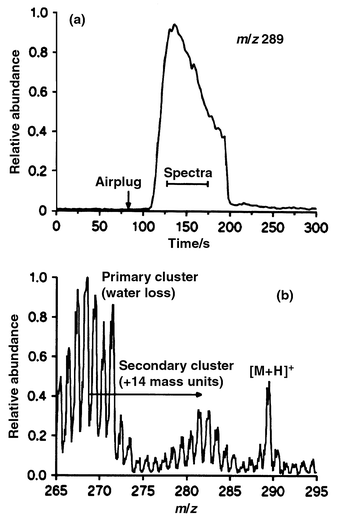 | ||
| Fig. 3 Single ion monitoring data (a) of the protonated molecule from testosterone obtained during the passage of a 90 s airplug and (b) desorption chemical ionization membrane inlet mass spectrum recorded as the avarage of 7 scans obtained during the desorption profile. | ||
A mass spectrum was also recorded during the desorption period. This spectrum, which is shown in Fig. 3(b), was recorded as an average of 7 spectra obtained during the desorption period [see Fig. 3(a)]. As expected the CI spectrum shows a significant ion at m/z 289 [M + H]+ from the protonated molecule. We were also expecting to see a large ion at m/z 271 [M + H − H2O]+ arising from water loss from the secondary alcohol. However, we observed a large cluster of ions from m/z 263–272. Similar clusters were observed at ±14 mass units around this cluster, but generally they were of lower intensity. Cholesterol, cholecalciferol and retinol gave the same set of cluster ions as testosterone, but they did not give the protonated molecule [M + H]+. In contrast, testosterone acetate and α-tocopherol gave no clusters and had a high intensity protonated molecule [M + H]+. The DCI-MIMS spectra did not change much with variation (0.1–0.4 Torr) in the CI gas pressure.
Normally, methane CI spectra of large hydrocarbons gives abundant [M − H]+ ions as a result of hydride ion abstraction and alcohols give abundant [M + H − H2O]+ ions. These expectations were confirmed by independently recorded methane CI spectra using a standard chemical ionization mass spectrometer (Finnigan MAT, San Jose, CA, USA, SSQ 710) and a solids probe. In the DCI-MIMS system described here many of the compounds tested gave a series of ion clusters separated by 14 mass units, the largest typically centred 20 mass units below the molecular ion. Apparently, the cluster formation is related the structure of the compound. Cholesterol, cholecalciferol and retinol, which contain a primary or secondary alcohol and a long hydrocarbon chain, only give the cluster ions, whereas testosterone acetate and α-tocopherol which do not contain a primary or secondary alcohol, give the protonated molecule but no ion clusters. Testosterone is somewhere in-between these two groups of compounds. It has the secondary alcohol but not the long hydrocarbon chain, just the steroid skeleton. In addition it has an extra oxygen in a cyclic ketone. We are unable to explain the chemistry/physics behind the formation of these cluster ions but speculate that they have something to do with a thermal decomposition (the membrane is heated to 300 °C during the desorption process). Clearly, the chemistry and physics behind the ion formation in the new DCI-MIMS system need further study.
Influence of the DCI-MIMS operation parameters
Two parameters, the sampling time and the membrane temperature, are particularly important for the DCI-MIMS system. Fig. 4 shows a plot of the area of the desorption peak as a function of sampling time. The data were obtained from a 20 μM retinol solution flushed through the membrane inlet with varying sampling times before the 90 s air plug was passed through the inlet. In this case the sample liquid was thermostated to 110 °C before it entered the ion source. The figure clearly shows that even a 30 min long sampling time is insufficient for the large biomolecules to diffuse into the membrane and establish a steady state between the number of analyte molecules entering the membrane and the number of analyte molecules leaving the membrane again, either back to the liquid sample or evaporating into the vacuum. The maximal amount of sample that can be collected in the membrane at a given concentration is therefore not reached within 30 min. The figure does show the beginnings of a saturation effect between 20 and 30 min and we chose 25 min as a standard sampling time in a compromise between sensitivity and acceptable sampling times.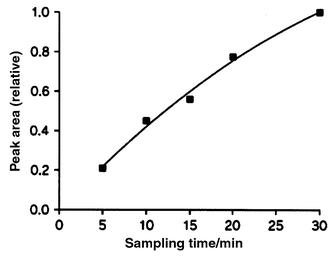 | ||
| Fig. 4 Area of the desorption profile from retinol (20 μM solution) as a function of sampling time. | ||
When the temperature of the sample liquid entering the ion source was increased (70–130 °C) we observed improved sensitivity and reduced memory effects. The effect was largest between 70 and 100 °C, where the area of the desorption peak increased by 80% and levelled off as the temperature approached 130 °C. In earlier work with trap-and-release MIMS6 we found that the sensitivity improved when the sample liquid was cooled. This was explained by an increased solubility of the sample molecules in the membrane with a lowering of the temperature. At that time the trap-and-release technique was used to detect semi-volatile organic compounds of a relatively small nature (for example phenoxyacetic acid). These ‘small’ compounds all reached a steady state permeation through the membrane within the 20 min sampling time used in contrast to the larger biomolecules studied here, which does not reach a steady state permeation through the membrane even at more than 100 °C. The large biomolecules simply diffuse so slowly in the membrane that they need time or elevated temperature to get into the membrane in sufficient amounts for analysis.
Quantitation
The DCI-MIMS system is particular sensitive to memory effects, since the membrane has two relatively cold spots (the attachments to the steel tubes). Analyte dissolved in the cold spots of the membrane does not evaporate into the mass spectrometer during the desorption period, but redistributes itself within the membrane as soon as the desorption period is over. In order to reduce the memory effect several cleaning airplugs (90 s duration) is passed through the system with pure water in-between. Table 1 demonstrates the efficiency of these cleaning air plugs. In these experiments the analysis procedure was: sample collection (retinol) 20 min, analysing air plug 90 s, pure water 60 s, cleaning air plug 90 s, repeated water/cleaning air plug sequences, blank sample 20 min, and finally a memory effect characterizing air plug 90 s. The table clearly shows that the memory effect after just one cleaning sequence is close to 25% and then it drops to 11 and 8% with 2 or 3 cleaning air plugs respectively. We therefore choose to use 3 cleaning air plugs, in-between all measurements to be sure that the memory effect was less than 10%.| Relative area of the desorption peak (m/z 268 monitored) | |||||
|---|---|---|---|---|---|
| Solution | Sample | 1st flush | 2nd flush | 3rd flush | Blank sample |
| A | 1.03 | 0.23 | 0.23 | ||
| B | 1.00 | 0.21 | 0.13 | 0.11 | |
| C | 0.94 | 0.25 | 0.13 | 0.08 | 0.08 |
Table 1 also gives a good impression of the reproducibility of the DCI-MIMS system. The three samples analysed were the same solution passed through the system with many cleaning air plugs in-between. As can be seen from the data the difference between the maximal and minimal values was less than 10%. In general we found that the reproducibility was better than 20% for all the compounds analysed.
Detection limits and linearity are also very important parameters of an analytical technique. These parameters are illustrated in Fig. 5, which shows single ion monitoring data of samples containing various concentrations of cholecalciferol (m/z 366 monitored). No signal was obtained from the blank sample, whereas the 2 μM sample gave a desorption peak in which the height was approximately 3 times the noise level. In the present work we have defined the detection limit as either the concentration that gives a desorption peak, which is 3 times higher than the noise level, or, in cases where there is a background peak (few ions coming from the membrane were observed), as the concentration that causes a doubling in peak area. Table 2 shows the measured detection limits for the six compounds tested. The detection limits varied from 10 μM for α-tocopherol and cholesterol to 0.2 μM for retinol and testosterone acetate. Two factors seem to influence the detection limits, the size of the molecule and the polarity. The large molecules have higher detection limits because they diffuse slower into the membrane during the sampling period and therefore are further away from the steady state concentration which gives the maximal signal (see Fig. 4). As usual with silicone membrane based systems polar compounds do not dissolve as well in the silicone membrane as do the non-polar compounds and the detection limits therefore increase with an increase in polarity. This is illustrated by a comparison of the detection limits for testosterone and testosterone acetate, where the esterification of the alcohol improved the detection limit by a factor of 5.
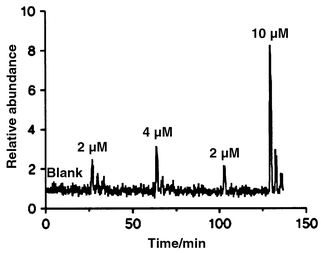 | ||
| Fig. 5 Single ion monitoring data (m/z 366 monitored) of low concentrations of cholecalciferol. | ||
| CI ions | |||||
|---|---|---|---|---|---|
| Compound | Mr | [MH]+ | Clusters | Ion monitored/ m/z | Detection limit/ μM |
| Retinol | 286 | No | Yes | 268 | 0.2 |
| Testosterone | 288 | Yes | Yes | 289 | 1 |
| Testosterone | |||||
| acetate | 330 | Yes | No | 331 | 0.2 |
| Cholecalciferol | 384 | No | Yes | 366 | 2 |
| Cholesterol | 386 | No | Yes | 368 | 10 |
| α-Tocopherol | 430 | Yes | No | 431 | 10 |
Fig. 5 also demonstrates the linearity of the method. The relative area of the desorption peaks was determined as zero, 2.0, 3.2, 2.1 and 10.7 for the zero, 2, 4, 2 and 10 μM samples, respectively. Although the 4 μM sample is a little low the system is obviously linear within the concentration range demonstrated here. The low 4 μM sample probably reflects the difficulties involved in the preparation of low level samples. In general, we found that the system was linear within 2 orders of magnitude.
Perspectives of the DCI-MIMS system
This work has clearly demonstrated that the DCI-MIMS system has potential as a method for the detection of large fat-soluble biomolecules directly in solution. In many cases characteristic protonated molecules ([M + H]+) are obtained, whereas in other cases the molecule decomposes as a result of the high membrane temperature during the desorption period. In the latter case a series of ion clusters appears separated by 14 mass units. The cluster formation makes unequivocal identification of compounds difficult and in its present form the method is best used for either a sample screening or quantitation of samples with known composition. The problem with the cluster production might be reduced if the system is rebuilt in a manner that gives a lower membrane temperature during the desorption period. At present the temperature inside the membrane reaches about 300 °C, which is too high for the large hydrocarbons. However, a lowering of the desorption temperature might also have a negative influence on the memory effects. Further, the DCI-MIMS system can be combined with tandem mass spectrometry and this will certainly assist in obtaining unequivocal identifications.The detection limits (around 1 μM) for the large biomolecules tested here are quite good considering the fact that this is the first MIMS system capable of detection of such compounds. However, they need to be improved by 2 orders of magnitude before our ultimate goal, the detection of steroid hormones directly in blood samples, is obtained. Our future research will therefore focus on an optimization of all parameters in the combined ion source–membrane interface, that is the geometry, the ion optics, the DCI chemistry and the physical operation parameters will be considered.
In an earlier paper13 we demonstrated that it is possible to detect highly polar, low molecular weight compounds with DCI-MIMS and a microporous polyacrylonitrile membrane. In contrast, the system described here uses a non-porous silicone membrane. With the silicone membrane the overall flux of both water and analyte into the mass spectrometer is about 5 orders of magnitude lower than with the porous polyacrylonitrile membrane. As a result of the membrane swap we have obtained a very efficient system for the detection of large fat-soluble biomolecules. However, we would still like to be able to detect the many polar biomolecules and we believe that this goal can be reached if a suitable membrane with properties between the silicone membrane and the polyacrylonitrile membrane can be found.
Acknowledgements
We thank Professor Jørgen Møller and Ole Tang Sørensen, Institute of Chemistry, SDU–Odense University, for useful discussions and for recording the standard methane CI spectra. Maria Anita Mendez thanks the CNQp, Brazil for financial support.References
- R. C. Johnson, N. Srinivasan and R. G. Cooks, Rapid Commun. Mass Spectrom., 1997, 11, 363 CrossRef CAS.
- S. Bohatka, Rapid Commun. Mass Spectrom., 1997, 11, 656 CrossRef CAS.
- F. R. Lauritsen and D. Lloyd, in Mass Spectrometry for the Characterization of Microorganisms, ed. C. Fenselau, ACS Symposium Series 541, American Chemical Society, Washington, DC, USA, 1994, pp. 91–106. Search PubMed.
- F. R. Lauritsen and T. Kotiaho, Reviews Anal. Chem., 1996, 4, 237 Search PubMed.
- N. Srinivasan, R. C. Johnson, N. Kasthurikrishnan, P. Wong and R. G. Cooks, Anal. Chim. Acta, 1997, 350, 257 CrossRef CAS.
- M. Leth and F. R. Lauritsen, Rapid Commun. Mass Spectrom., 1995, 9, 591 CAS.
- F. R. Lauritsen and R. A. Ketola, Anal. Chem., 1997, 69, 4917 CrossRef CAS.
- T. Kotiaho, J. Mass Spectrom., 1996, 31, 1 CrossRef CAS.
- G. Matz and P. Kesners, Anal. Mag., 1995, 23, M12 Search PubMed.
- M. H. Soni, J. Callahan and S. McElvany, Anal. Chem., 1998, 70, 3103 CrossRef CAS.
- M. H. Soni, A. P. Baronavski and S. McElvany, Rapid Commun. Mass Spectrom., 1998, 12, 1635 CrossRef CAS.
- F. R. Lauritsen, T. K. Choudhury, L. E. Dejarme and R. G. Cooks, Anal. Chim. Acta, 1992, 266, 1 CrossRef CAS.
- R. A. Ketola and F. R. Lauritsen, Rapid Commun. Mass Spectrom., 1999, 13, 749 CrossRef CAS.
- M. A. Baldwin and F. C. McLafferty, Org. Mass Spectrom., 1973, 7, 1353 Search PubMed.
Footnote |
| † Present address: State University of Campinas, Institute of Chemistry, CP 6154, 13083-970 Campinas, SP, Brazil. |
| This journal is © The Royal Society of Chemistry 2000 |