Reagentless enzyme electrode based on phenothiazine mediation of horseradish peroxidase for subnanomolar hydrogen peroxide determination†
Silvia Serradilla Razola, Esra Aktas, Jean-Claude Viré and Jean-Michel Kauffmann*
Institute of Pharmacy, Université Libre de Bruxelles, Campus Plaine CP 205/6, 1050, Bruxelles, Belgium
First published on UnassignedUnassigned7th January 2000
Abstract
The development and characterization of a highly sensitive enzyme immobilized carbon based electrode for the determination of subnanomolar concentrations of hydrogen peroxide in aqueous samples is described. The biosensor consists of horseradish peroxidase (HRP) immobilized in solid carbon paste along with a suitable redox mediator. The latter allows the acceleration of the electroreduction of HRP in the presence of hydrogen peroxide. Several phenothiazines as mediators are investigated in a comparative manner and with respect to dimethylferrocene using cyclic voltammetry and amperometry. Insolubilization of the HRP in the solid carbon paste is achieved by cross-linking the enzyme with glutaraldehyde and bovine serum albumin. Several experimental parameters such as pH, mediator and enzyme content are considered. The hydrogen peroxide determination is better carried out in 0.1 M acetate buffer, pH 4.5, by amperometry at an applied potential of 0.0 V versus Ag/AgCl, 3 M NaCl concentration and by using the phenothiazine base as redox mediator. The biosensor response is linear over the concentration range 2 nM–10 μM with a detection limit of 1 nM. The linear range of the hydrogen peroxide response without a mediator in the biosensor is found between 2 and 40 μM. The biosensor can be used for more than 180 measurements. Additional modification of the electrode by incorporation of Nafion SAC-13 microparticles in the solid carbon paste allows detection of concentrations of hydrogen peroxide as low as 0.1 nM.
Introduction
Hydrogen peroxide is probably one of the most extensively investigated molecules with a variety of techniques and in many different domains such as in clinical, food, pharmaceutical and environmental analyses.1 In several circumstances, extremely low amounts of hydrogen peroxide, i.e., nanomolar concentrations, must be determined, such as in marine water,2,3 air,3,4 drinking water,5 at the level of a single cell in vitro6 or in vivo during oxidative stress7 in food samples and in many immunoassays.8 Because of its high instability and because of the complexity of the samples to be analysed, analytical techniques allowing accurate and fast determinations of very low concentrations are scarce.Chemiluminescent,3,9 fluorimetric2,10 and electrochemical11,12 methods are the most sensitive for hydrogen peroxide. The last two exploit advantageously an enzymatically catalysed reaction between hydrogen peroxide and an appropriate substrate, with subsequent change of the fluorescence intensity or acceleration of the electron transfer rate.
HRP has been most thoroughly studied and frequently used to exemplify the peroxidase reaction cycle:
Native
peroxidase + H2O2
→ Compound I +
H2O (Fe3+)
(Fe4+
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O,
P+)Compound I + AH2
→ Compound II +
AH* (Fe4+
O,
P+) (Fe4+
O)Compound II + AH2
→ Native peroxidase + AH*
H2O (Fe4+
O) (Fe3+) O,
P+)Compound I + AH2
→ Compound II +
AH* (Fe4+
O,
P+) (Fe4+
O)Compound II + AH2
→ Native peroxidase + AH*
H2O (Fe4+
O) (Fe3+) | (1) |
Particularly suitable and often used mediators are ferrocene and its derivatives,16–20 osmium based polymers,21,22 and conducting organic salts such as tetrathiafulvalene and tetracyano-p-quinodimethane.23 The mediators must have access to the enzyme redox centre and allow fast electron transfer to the electrode surface. Biosensors with the mediator immobilized must allow facile diffusion of the redox species between the enzyme and the electrode or must facilitate electron transport through an electron hopping phenomenon as obtained by the use of osmium redox polymers.21,22 Carbon based electrodes are generally employed for HRP biosensors provided that the electrode material is activated for allowing fast electron transfer to occur.13 Especially attractive are carbon composites, since the enzyme and the mediator may be readily retained in the electrode matrix.24 In the blend, both the biocomponent and the mediator are in intimate contact and located in the vicinity of the electrode surface (carbon particles). We have recently shown that a solid paraffin–graphite matrix offers robustness and good analytical characteristics.25,26 This structure allows the biocomponent to be readily immobilised by dispersion of the enzyme caused by heating slightly above the melting point of solid paraffin25,26 or by the use of an appropriate organic solvent.27 In the present article, immobilization of both the enzyme horseradish peroxidase and the mediator was achieved in a solid carbon paste electrode (sCPE) with the hope that the mediation efficiency would be retained in the solid matrix. Additional innovation consists of the use of the phenothiazine base as redox mediator. Phenothiazine type molecules, such as methylene blue,28,29 methylene green30 thionine31,32 or toluidine blue,27 have already been used with HRP-electrodes, but these molecules are soluble in water, exhibit a distinct electrochemical pattern compared with phenothiazine base and must be immobilized (physically or chemically) to be retained in the biosensor. Recently, L. Gorton and his group secured mediator immobilization by developing a HRP carbon paste containing an acrylamide polymer with linked toluidine blue for hydrogen peroxide determination with a detection limit of 50 nM.27 L. Deng et al. published a series of similar articles on peroxidase biosensors containing phenothiazine-like mediators28–30 and recently suggested methylene green with HRP retention in zeolite particles.33 Previously, we have shown that phenothiazine drugs can be determined amperometrically down to 10 nM using a HRP modified sCPE in the presence of hydrogen peroxide.34 The electrochemistry of these molecules is well known35,36 and the HRP oxidation of phenothiazines has been extensively investigated.37–40 It appears that the oxidation, both enzymatic and electrochemical, gives rise to a relatively stable cation radical. The latter exhibits reversible electrochemical behaviour at solid electrodes at low potentials in acidic and non-aqueous media. This prompted us to investigate comparatively several phenothiazines as possible redox mediators for HRP for hydrogen peroxide determination.
Experimental
Apparatus
Amperometric measurements were obtained with a conventional three-electrode cell using a BAS working electrode (active geometric area = 3 mm), a Ag/AgCl 3 M NaCl reference electrode and a platinum wire as counter electrode. The solution (10 mL) was stirred with a magnetic bar at approximately 500 rpm. The potentiostat was a Brüker E-230 LC (Brüker, Brussels, Belgium). The current output was displayed on a Y/t Kipp and Zonen recorder. Cyclic voltammetry was performed using a BAS potentiostat Model CV 27 (Bioanalytical Systems Ltd., West Lafayette, IN, USA) connected to a 7090 HP recorder (Hewlett Packard, Brussels, Belgium). All experiments were made at room temperature (22 ± 1 °C).Reagents and solutions
All reagents were of analytical grade and the solutions were prepared with Milli-Q quality water. Buffer solutions were prepared from sodium acetate (Vel, Leuven, Belgium) and adjusted to pH by acetic acid (Merck, Overijse, Belgium). Phenothiazine base (PhZ b), promazine HCl (PMZ, HCl), promethazine HCl (PmZ, HCl), thioridazine HCl , peroxidase from horseradish (E.C., 1. 11.1.7, 170 U per mg, type II) (HRP), 1,1′-dimethylferrocene (1,1′ dmFc) and fumed silica were from Sigma (Bornem, Belgium). Promethazine base (PmZ b) was obtained from its chlorhydrate derivative by precipitation in alkaline media, filtration and desiccation under vacuum. Bovine serum albumin (BSA), Nafion SAC-13 and Nafion 117 solution in lower aliphatic alcohols were from Aldrich (Bornem, Belgium). Solid paraffin, glycine and glutaraldehyde, 25% aqueous solution (GA), were from Merck. Graphite powder was obtained from Connex (Conmetal, Celle, Belgium).Hydrogen peroxide was from Acros (Geel, Belgium) and assayed by volumetric titration with a permanganate solution.Graphite pretreatment
Graphite particles were first cleaned with acetone, then rinsed with water and finally activated with aqua regia for 2 h. Finally, the particles were washed with deionized water until neutral pH of the filtrate is obtained. The graphite was dried at 400 °C during 2 h before use.Enzyme insolubilisation
The cross-linking of the HRP to the BSA was achieved by reaction with glutaraldehyde (2.5% aqueous solution). This was carried out on a glass surface by use of gentle stirring, with a thin glass rod, of HRP (3.33% m/m) and BSA (1.66% m/m) in 50 μL of 0.1 mmol L−1 acetate buffer and then adding 50 μL of GA. The mix was left at 5 °C for 1 h. Finally, excess GA in the insoluble biocomponent was thoroughly reacted by rinsing with 0.1 mmol glycine solution in acetate buffer. It was left to dry at room temperature. The crystals formed were scraped off with a spatula and their size was homogenized in a mortar using a pestle.Preparation of the enzyme modified solid carbon paste electrode (HRP–sCPE)
The HRP immobilized solid carbon paste (HRP–sCP) was obtained first by melting the paraffin wax (33% m/m) in a mortar dipped in a water-bath at 50 °C. With the mortar outside the bath, the HRP–GA–BSA powder (5% m/m) was dispersed in the wax by thorough mixing with a glass spatula. The graphite particles (62% m/m) were promptly incorporated to this viscous paste by mixing. The paste slowly solidified at room temperature, then it was crushed to a homogeneous consistency. This paste was pressed into the well of the electrode body. The surface of the HRP–sCPE was polished manually on a soft clean paper before use.Mediator modified HRP–sCPE
The mediator was dissolved in the minimum amount of the appropriate solvent (MQ-water or diethyl ether) and mixed with graphite. The powder was ready for use after solvent evaporation at room temperature. Then the graphite with adsorbed mediator was added to the mix formed by the paraffin wax and HRP–BSA as above.Nafion modified biosensor
The biosensors were prepared as above except that the graphite was previously modified by Nafion in two different ways: (a) Nafion SAC-13 was mixed with graphite by grinding in a mortar, then the resulting powder was homogenized using a vortex mixer; (b) Nafion in ethanol (0.5% m/v) was mixed with graphite and the solvent allowed to evaporate in air at ambient temperature.Electrode pretreatment
Before the experiments the biosensor was left for 5 min in a stirred acetate buffer solution, pH 4.5, to wash out any loosely adsorbed mediator and/or enzyme. A longer duration of preconditioning gave no different behaviour. After preconditioning, the biosensor was removed, rinsed with water and dipped into the measuring cell. This step appeared to be mandatory for obtaining reproducible of the results.Results and discussion
Enzyme immobilization
The enzyme HRP is a small protein that is highly water soluble and it may readily leak out of the electrode matrix.27 Modification of the enzyme to render it less water soluble may be achieved by different methods, viz., cross-linking with GA in the presence of an inert protein such as BSA28,31 oxidation with periodate27 or lipophilizing the enzyme by the use of caprylic aldehyde.23 Here, we applied the glutaraldehyde–BSA cross-linking procedure, and the calibration curve obtained for the amperometric determination of hydrogen peroxide in the concentration range 0.01–90 μmol L−1 was lower for HRP plus phenothiazine with a slope of about 75% compared with the HRP–GA–BSA plus phenothiazine biosensor. The lower response at the former can be attributed to leaking of the HRP into the solution during 5 min of preconditioning, as demonstrated previously27,41Influence of the amount of biocomponent in the electrode matrix
This study was performed with biosensors containing 3, 5 or 7.5% of biocomponent (HRP–GA–BSA) in the paste in order to determine the maximum amount of enzyme for high sensitivity while keeping the mechanical integrity of the paste. The latter may be affected by a too high amount of proteins leading to swelling phenomena at the electrode–water interface and facile leaking of the biocomponent and mediator into the aqueous solution. Calibration curves created in the concentration range of 0.01–2 μmol L−1 showed a slope about 50% higher with the 5% content than with the 3% protein content, while higher ratios (7.5%) gave the lowest sensitivity. This can again be related to biocomponent washing out during the preconditioning of the paste. Note that leaking of the mediator out of the biosensor during the pretreatment (see below) is supposed not to be influenced by changes in the protein content.Influence of the mediator in the paste
Cyclic voltammograms obtained in acetate buffer pH 4.5 with the sCPE containing 2% phenothiazine showed the quasi-reversible behaviour of the mediator: EpEpc = 350 mV versus Ag/AgCl, NaCl 3 M. The peak potential remained unchanged and the response was controlled by diffusion of the mediator, as inferred from the linearity between Ip/v1/2 in the scan range investigated, i.e., 5–100 mV s−1. The redox process corresponds to the removal of one electron, i.e., formation of the corresponding cation radical. Addition of hydrogen peroxide had no influence on the cyclic voltammetry redox peaks and, in amperometry with the electrode poised at 0 V, no response was observed when varying the hydrogen peroxide concentration from 0.01 to 1 μM.In the presence of the enzyme in the paste (5% HRP–GA–BSA), the cyclic voltammetric response of the phenothiazine base (2% m/m in the paste) increased approximately three-fold, while the dimethylferrocene oxidation current rose slightly and the promethazine base oxidation current intensity dropped by approximately 30%. After pretreatment, the cyclic voltammogram showed a 40% peak current drop for the phenothiazines and more than 60% for dimethylferrocene by comparing with the cyclic voltammogram before stirring. A loss of weakly adsorbed mediator was inferred. This mediator wash out occurred essentially during the first 5 min of stirring, after which the voltammogram stabilized; such behaviour has already been reported.42 Following biosensor preconditioning, the cyclic voltammogram showed no significant changes in the redox curve shape (potential and intensity) except in the case of the promethazine entrapped biosensor. Indeed, repetitive scanning at the latter showed a slight but progressive diminution of the cation radical redox peak currents, with formation of a new redox couple located at less positive potentials (Epa = +210 mV, Epc = +180 mV) (Fig. 1). This suggests a more complex pattern involved in the oxidation peak of promethazine with some relatively fast chemical reactions occurring subsequent to promethazine cation radical formation. This gives rise to species more readily oxidized than the parent compound. This new redox peak was also clearly observed at the solid carbon paste electrode when studying promethazine hydrochloride electrooxidation in solution and when studying its oxidative transformation by HRP in solution in the presence of hydrogen peroxide.41 The mechanism of promethazine oxidation is rather complex, giving rise to several intermediates and oxidized compounds.43 While not specifically identified in the literature, we may postulate that the new redox couple probably corresponds to a promethazine hydroxylated derivative (see also ref. 35). No new redox couples have been observed for the phenothiazine base entrapped in the carbon paste, at least during the time scale of the voltammetric experiments. By addition of hydrogen peroxide to the solution, a considerable increase in the cathodic current was obtained in cyclic voltammetry, especially for phenothiazine and promethazine bases (Fig. 2, A and B). This dramatic increase in the reduction current is a result of the regeneration of the cation radical by the enzyme at the electrode interface. The shape of the voltammogram, in the presence of hydrogen peroxide above 0.5 mM, was characteristic of a catalytic process with no corresponding oxidation current, indicating that all the electroactive molecules at the electrode interface are efficiently mediating electrons between the HRP molecules and the graphite particles. At lower concentrations of hydrogen peroxide catalysis was observed but oxidation of the mediator was still apparent in the voltammogram (not shown). For the dimethylferrocene biosensor, the efficiency of the catalysis was less high and the anodic portion of the voltammogram was still detected (Fig. 2, C).
 | ||
| Fig. 1 Cyclic voltammogram in 0.1 M acetate buffer, pH 4.5, at the enzyme electrode. sCPE modified by 2% PMZ b + 5% HRP–GA–BSA. Voltammogram recorded after several cycles. Scan rate 25 mV s−1, 5 min preconditioning. | ||
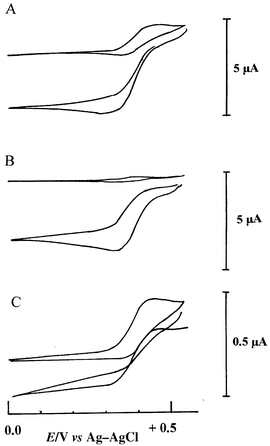 | ||
| Fig. 2 Cyclic voltammograms in 0.1 M acetate buffer, pH 4.5, at the enzyme electrodes: A, modified by PhZ b; B, modified by PMZ b; C, modified by dmFc ; (a) upper traces, in the absence of hydrogen peroxide; (b) lower traces, in the presence of 0.5 mmol L−1 hydrogen peroxide. Scan rate 5 mV s−1. 0.1 mol L –1 acetate buffer, pH 4.5. | ||
Amperometric measurements of hydrogen peroxide, in the concentration range 0.08–0.8 μM, at the different biosensors (with different mediators) were compared with the biosensor without mediator. As is shown in Fig. 3, the biosensors containing a water soluble phenothiazine (hydrochloride derivative) gave less sensitive responses (because of mediator readily leaching out during the preconditioning). The biosensors with the hydrophobic species showed the highest response. With dimethylferrocene as mediator, the slope of the response was two-fold lower than with phenothiazine and three-fold lower than with promethazine. In the absence of mediator in the paste (curve not shown), the direct reduction of the oxidized form of the enzyme (HRP compound 1) was only detected at a concentration of hydrogen peroxide above 0.8 μM with a slope twenty-fold lower than the phenothiazine base biosensor.
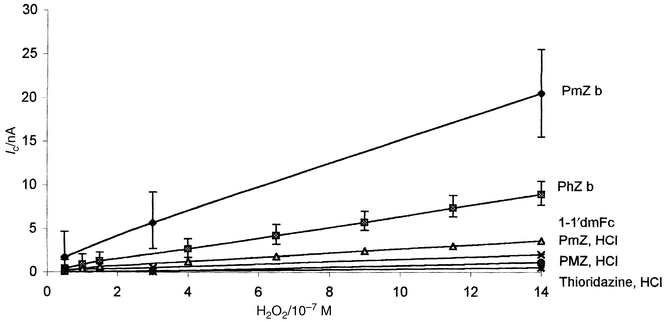 | ||
| Fig. 3 Influence of the nature of the mediator on the biosensor response. Amperometric calibration curves at the sCPE modified by 2% mediator and 5% HRP–GA–BSA. Acetate buffer pH 4.5. Error bars only for PmZb and PhZb curves for figure clarity. | ||
From these observations, and taking into account literature data on HRP biosensors mediated by ferrocene derivatives16–20 and on HRP oxidation of phenothiazines,37–40 we may postulate that the current recorded at the present biosensor corresponded to the electroreduction of the cation radical of the phenothiazine. Based on the formal redox potential of HRP compound I and compound II in slightly acidic media (both equal to approximately 0.9 V versus NHE),18 two phenothiazine molecules may react with one HRP (Fig. 4). In the case of promethazine base as mediator, an additional contribution of the reduction of promethazin-3-one species may be inferred. The latter may explain the higher sensitivity of the promethazine base calibration curve. However, the biosensor with promethazine gave a higher detection limit (S/N = 3) than for the phenothiazine based biosensor because of the higher baseline noise and lower reproducibility at the former (see error bars in the calibration curve of PmZb compared with PhZb in Fig. 3). This could be related to the progressive leaking of promethazine oxidized species and to the solubility of promethazine base attributed to its dimethylaminopropyl side chain (pKa = 9.1) into the acidic buffer comparing to phenothiazine base (pKa = 2.5).
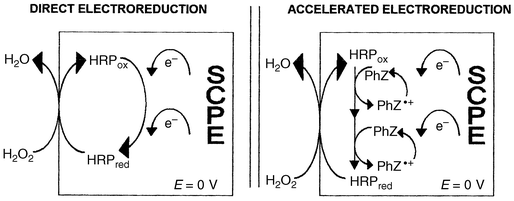 | ||
| Fig. 4 Schematic mechanism of direct and mediated bioelectrocatalytic reduction of hydrogen peroxide at the HRP modifed sCPE. | ||
By increasing the phenothiazine base loading, from 0.5 to 1%, the biocatalytic current was raised considerably, and higher loadings (up to 3%) showed no substantial improvement. The KMapp, as estimated from Imax/2, for a 2% m/m immobilized biosensor, was 15 μmol L−1. Further experiments were carried out with a phenothiazine loading of 2% m/m, which corresponded to the current saturation value.
Interferences
Many compounds may interfere at the electrode surface, despite the low applied potential, because HRP has a broad substrate activity giving rise to oxidized compounds which may be reduced at the applied potential.44–46 Species such as ascorbic acid may interact with the enzymatically generated radical;47–49 others may inhibit the enzyme activity by interacting with the heme active centre of the enzyme, such as cyanide,50 and proteins and lipids in the analysed sample may foul the electrode surface.Several strategies can be applied for minimizing interferences, e.g., charge repulsion (in the case of ascorbate and urate), involving coating the biosensor surface with a negatively charged membrane such as Nafion.51 We have studied the response of two distinct Nafion modified biosensors with respect to the hydrogen peroxide response (see Experimental section). Nafion-SAC 13 consists of silica nanoparticles coated with Nafion and was dispersed into the sCP matrix, and Nafion 117 polymer dissolved in ethanol was coated onto graphite before paste preparation. The calibration curves were obtained by amperometry, using the standard addition method, as a function of hydrogen peroxide in the concentration range 0.01–0.33 μmol L−1. The highest slopes of the calibration curves were obtained at the Nafion-SAC 13 based biosensor. The biosensor without Nafion gave a response corresponding to 70% of Nafion SAC-13 biosensor and the lowest slope was obtained with Nafion 117, with a slope equal to 18% of Nafion SAC-13 biosensor. The lower sensitivity at the latter probably resulted because of the homogeneous coating of the graphite particles with the polymer, which constituted a diffusional barrier for the phenothiazine. Nafion SAC-13 mixed into the paste as solid particles (1% m/m) offered signal improvement, a phenomenon not well understood yet but already reported for silica based CPEs.52 Some swelling phenomenon (visually detected) due the sulfonated functional groups was inferred. This swelling process probably increased the biosensor active area, and a sub-nanomolar concentration of hydrogen peroxide could be detected. Since the biosensor with Nafion SAC-13 rapidly deteriorated on successive use, the loading of Nafion SAC-13 was varied from 1 to 10%. As is shown in Fig. 5, the sensitivity of the biosensor increased with the amount of SAC-13 in the sCPE, but substantial surface swelling occurred at higher ratio.
 | ||
| Fig. 5 Amperometric calibration curves at the sCPE-PhZb (2%) HRP–GA–BSA (5%) as a function of Nafion SAC-13 content: A, 10%, B, 5%; C, 1%, D, without Nafion SAC-13. Acetate buffer, 0.1 M pH 4.5. | ||
Operational stability
This was checked comparatively at the HRP–GA–BSA-phenothiazine biosensor and at the same biosensor modified by Nafion 117 or SAC-13 Nafion powder, by performing daily a calibration curve between 0.01–50 μM in acetate buffer, pH 4.5. The electrode was stored dry at 5 °C when not in use. As is shown in Table 1, after 24 h, the biosensor sensitivity dropped to only 33% of its original value at the HRP–GA–BSA sCPE, while in the presence of Nafion the response decline was lowered. The latter might be related to the swelling phenomenon at the Nafion based biosensor, which compensates for the biocomponent activity decrease. Better stabilities of HRP-based biosensor may be achieved by applying other immobilization procedures or by using additional stabilizers in the electrode.15| Response | |||
|---|---|---|---|
| (A) HRP–PhZ sCP | (B) A + 1% NAFION 117 | (C) A + 1% NAFION SAC-13 | |
| First use | 100% | 100% | 100% |
| +24 h | 33% | 88% | 67% |
| +48 h | 2% | 75% | 20% |
| +72 h | 65% | 22% | |
| +96 h | 43% | 20% | |
| +120 h | 33% | 15% | |
| +144 h | 25% | 9% | |
Using the same HRP–GA–BSA phenothiazine biosensor surface (just rinsing the probe after each calibration curve),180 measurements have been made in a working day period (8 h), and the reproducibility of the slope of 30 calibration curves, in the concentration range 0.5–70 μM, gave an RSD of 6.7%. The latter stability data were better than those shown in Table 1, i.e., it is better to use the biosensor continuously than to keep it dry at low temperature after one assay. At this stage no interpretations could be provided regarding stability data, and it is recommended to renew the biosensor surface after one day of use.
Taking into account the above results, and considering that the biosensor composition should be adjusted depending on its application, further experiments consisted in characterizing the biosensor in acetate buffer, pH 4.5, with a paste composition of 3.33% HRP cross-linked with 1.66% BSA, 2% phenothiazine base, 32% solid paraffin and 61% graphite powder.
Influence of pH and applied potential
The biosensor was tested by performing calibration curves for hydrogen peroxide at different potentials in the range between −0.2 and +0.2 V. The highest signal was obtained at −0.1 V, stabilization of baseline occurred within 2 min, but the background current was higher and the reproducibility of the results less than at 0 V. At the latter, a steady-state baseline current was achieved, typically within 1 min and for the different concentrations investigated.The amperometric response of a 10 μM solution of hydrogen peroxide was investigated in 0.1 M acetate buffer in the pH range 3.5–6. As is shown in Fig. 6, higher responses were observed in the pH range 4.5–6. The former was selected because of its buffering power and better reproducibility of the results (RSD = 2.5, n = 4). We may also recall that the phenothiazine cation radical is more stable in acidic media and in a hydrophobic environment.53
 | ||
| Fig. 6 Influence of pH on the biosensor (sCPE-PhZb 2 %-HRP–GA–BSA 5%) response. Acetate buffer 0.1 M, hydrogen peroxide 0.3 μmol L−1. | ||
Some applications in clinical and pharmacological practice require hydrogen peroxide determination in neutral pH. The biosensor response was tested in 0.1 M phosphate buffer, pH 7.4. A longer stabilization of the baseline current was observed (five-fold longer than at pH 4.5) and the background noise was slightly higher. The slope of the hydrogen peroxide calibration curve studied in the concentration range 0.5–70 μM was identical (within experimental error) than at pH 4.5 but an important decrease of the response was observed in a daytime use with the same surface.
Analytical characteristics
The linear range for hydrogen peroxide was determined amperometrically by the standard additions method in 0.1 M acetate buffer at both mediated and non-mediated biosensors with an applied potential of 0 V. The response time was very rapid with 90% of the steady state signal achieved within 6 and 3 s for 1 μM and 0.01 μM hydrogen peroxide concentrations, respectively. A typical current–time profile for stepwise increases of nanomolar concentrations of hydrogen peroxide is depicted in Fig. 7. The phenothiazine based biosensor allowed quantifications over a very broad concentration range and down to 2 nmol L−1. The quantification limit was 3 orders of magnitude higher at the non-mediated biosensor; a linear relationship was obtained between 1.8 and 40 μM [y (nA) = 1.67 × 105 (M) x + 0.34, r2 = 0.980] at the HRP–BSA sCPE and between 2 nM and 40 μM [y (nA) = 9 × 106 (M) x + 0.19, r2 = 0.999] at the same electrode modified by 2% m/m PhZb. The detection limits (S/N = 3) were 0.8 μM, 1 nM and 0.1 nM at the HRP–BSA, HRP–BSA plus PhZb (2%) and HRP–BSA plus PhZb (2%) plus Nafion Sac-13 (5%) sCPEs, respectively. | ||
| Fig. 7 Typical amperometric responses in acetate buffer pH 4.5, for step additions of hydrogen peroxide at the biosensors described in Fig 5. Final concentration of hydrogen peroxide: 5, 30, 80, 180, 380, 780, 980 nmol L−1. | ||
Storage stability of the HRP–GA–BSA, PhZ, solid carbon paste
The storage stability was determined by keeping the mediator based HRP–BSA paste at 5 °C. The slope of the calibration response for hydrogen peroxide, in the 0.01–0.1 μmol l−1 range, dropped by about 1.5% after 4 months.Conclusion
The use of phenothiazine base as a redox mediator for the enzyme horseradish peroxidase in a solid carbon paste electrode offers very high sensitivity for hydrogen peroxide detection. The phenothiazine molecule is practically insoluble in water and exhibits a reversible one electron transfer electrochemical behaviour in the solid carbon paste. In the presence of hydrogen peroxide, the enzymatic oxidation of immobilized phenothiazine leads to its cation radical, which is reduced at a potential close to 0 V versus Ag/AgCl. The efficiency of the redox mediation, observed in cyclic voltammetry and illustrated by the broad quantification domain down to 2 nM, may be related to the excellent charge and electron transfer capabilities of the phenothiazine molecule in biological systems and at carbon electrodes54 and its high interaction with protein species.55 Phenothiazine is light sensitive but the shelf life of the phenothiazine based HRP–GA–BSA paste biosensor was higher than 4 months. This biosensor configuration should be attractive for trace determinations of hydrogen peroxide requiring short time resolution and for biosensor development comprising additional immobilized oxidases and, eventually, permselective membranes. Alternatively, other strategies for enzyme and mediator immobilization in the paste can be considered, such as retention in zeolite particles.33Acknowledgements
Thanks are expressed to the ‘Communauté Française de Belgique’ and to the ‘Ministère des Affaires Etrangères Espagnoles’ for financial support to S.S.R.References
- C.-L. Wang and A. Mulchandani, Anal. Chem., 1995, 67, 1109 CrossRef CAS.
- L.-S. Zhang and G. T. F. Wong, Talanta, 1999, 48, 1031 CrossRef CAS.
- D. Price, R. Fauzi, C. Mantoura and P. J. Worsfold, Anal. Chim. Acta, 1998, 371, 205 CrossRef CAS.
- J. Yuan and A. M. Shiller, Anal. Chem., 1999, 71, 1975 CrossRef CAS.
- J.-M. Lin, H. Arakawa and H. Yamada, Anal. Chim. Acta, 1998, 371, 171 CrossRef CAS.
- C. E. B. Almeida, D. L. Felicio, R. S. Galhardo, J. B. Cabral-Neto and A. C. Leitao, Mutat. Res., 1999, 433, 59 CrossRef CAS.
- Y. Matsui and Y. Kumugae, Neurosci. Lett., 1991, 126, 175 CrossRef CAS.
- J. Zhang and Z. Ling, Talanta, 1994, 44, 1999.
- M. Arnold, X. Zhou and R. S. Petsch, Talanta, 1994, 41, 783 CrossRef CAS.
- Y.-Z. Li and A. Townshend, Anal. Chim. Acta, 1998, 359, 149 CrossRef CAS.
- J. E. Frew, M. A. H. Harmer, H. A. O. Hill and S. I. Libor, J. Electroanal. Chem., 1986, 201, 1 CrossRef CAS.
- V. A. Bogdanovskaya, V. A. Fridman, M. R . Tarasevich and F. Scheller, Anal. Lett., 1994, 27, 2823 CAS.
- L. Gorton, G. Jönsson, E. Petterson, E. Csöregi, K. Johansson, E. Dominguez and G. Marko-Varga, Analyst, 1992, 117, 1235 RSC.
- H. Kinoshita, M. Torimura, K. Kano and T. Ikeda, Electroanalysis, 1997, 9, 1234 CAS.
- T. Ruzgas, E. Csöregi, J. Emneus, L. Gorton and G. Marko-Varga, Anal. Chim. Acta, 1996, 330, 123 CrossRef CAS.
- A. L. Ghindilis, P. Atanasov and E. Wilkins, Electroanalysis, 1997, 9, 661 CAS.
- A. J. Reviejo, F. Liu, J. M. Pingarron and J. Wang, J. Electroanal. Chem., 1994, 374, 133 CrossRef CAS.
- S. J. Sadeghi, G. Gilardi and A. E. G. Cass, Biosens. Bioelectron., 1997, 12, 1191 CrossRef CAS.
- S. L. Chut, J. Li and S. N. Tan, Analyst, 1997, 122, 1431 RSC.
- M. A. Del Cerro, G. Cayuela, A. J. Reviejo, J. M. Pingarron and J. Wang, Electroanalysis, 1997, 9, 1113 CAS.
- A. Gregg and A. Heller, Anal. Chem., 1990, 62, 258 CrossRef.
- M. C. Garguilo, N. Huynh, A. Proctor and A. C. Michael, Anal. Chem., 1993, 65, 523 CrossRef CAS.
- U. Korell and U. E. Spichiger, Anal. Chem., 1994, 66, 510 CrossRef CAS.
- U. Wollenberger, J. Wang, M. Ozsoz and E. Gonzalez-Romero, Bioelectrochem. Bioenerg., 1991, 26, 287 CrossRef CAS.
- C. Petit, A. Gonzales-Cortes and J.-M. Kauffmann, Talanta, 1995, 42, 1783 CrossRef CAS.
- M. Pravda, C. Petit, Y. Michotte, J.-M. Kauffmann and K. Vytras, J. Chromatogr., 1996, 727, 47 CrossRef CAS.
- V. Rajendran, E. Csöregi, Y. Okamoto and L. Gorton, Anal. Chim. Acta, 1998, 373, 241 CrossRef CAS.
- C. Lei, Z. Zhang, H. Liu, J. Kong and J. Deng, Anal. Chim. Acta, 1996, 332, 73 CrossRef CAS.
- J. Qian, Y. Liu, H. Liu, H. Yu and J. Deng, Anal. Biochem., 1996, 236, 208 CrossRef CAS.
- H. Liu, Y. Liu, J. Qian, J. Yu and J. Deng, Talanta, 1996, 43, 111 CrossRef CAS.
- J. -J Xu, D. -M Zhou and H. -Y Chen, Electroanalysis, 1998, 10, 713 CrossRef.
- C. Ruan, F. Yang, C. Lei and J. Deng, Anal. Chem., 1998, 70, 1721 CrossRef CAS.
- B. Liu, F. Y. Kong and J. Deng, Anal. Chim. Acta, 1999, 386, 31 CrossRef CAS.
- C. Petit, K. Murakami, A. Quilinc, J.-F. Liegeois and J.-M. Kauffmann, Electroanalysis, 1998, 10, 1241 CrossRef CAS.
- M. Neptune and R. L. McCreery, J. Org. Chem., 1978, 43, 5006 CrossRef CAS.
- W. J. M. Underberg, J. Pharm. Sci., 1978, 67, 1131 CAS.
- A. Vazquez, J. Tudela, R. Varon and F. Garcia Canovas, Anal. Biochem., 1992, 202, 245 CrossRef CAS.
- M. Nakano, K. Sugioka, H. Nakano, C. Takyu and H. Inaba, Biomed. Biophys. Res. Commun., 1985, 130, 952 Search PubMed.
- A. Vazquez, J. Tudela, R. Varon and R. Garcia-Canovas, Biochem. Pharmacol., 1992, 44, 889 CrossRef CAS.
- J. Escribano, F. Garcia-Canovas, F. Garcia-Carmona and J. A. Lozano, Biochim. Biophys. Acta, 1985, 831, 313 CAS.
- F. Van Gijseghem, J. Pinto, S. Serradilla Razola and J.-M. Kauffmann, submitted for publication..
- U. Wollenberger, A. Drungiliene, W. Stöcklein, J. J. Kulys and F. W. Scheller, Anal. Chim. Acta, 1996, 329, 231 CrossRef CAS.
- W. J. Underberg, J. Pharm. Sci., 1978, 65, 1133.
- G. Meunier, J. De Montauzon, J. Bernadou, G. Grassy, M. Bonnafous, S. Cros and B. Meunier, Mol. Pharmacol., 1998, 33, 93 Search PubMed.
- T. Ruzgas, J. Emneus, L. Gorton and G. Marko-Varga, Anal. Chim. Acta, 1995, 311, 245 CrossRef CAS.
- F. Dayon, Arch. Biochem. Biophys., 1998, 351, 27 CrossRef.
- W. L. Baker, Anal. Lett., 1998, 31, 1325 CAS.
- F. E. Dyan, S. O. Duke, V. Faibis, F. M. Jacobs and N. J. Jacobs, Arch. Biochem. Biophys., 1998, 351, 27 CrossRef.
- D. C. Goodwin, I. Yamazaki, S. D. Aust and T. A. Grover, Anal. Biochem., 1995, 231, 333 CrossRef CAS.
- T.-M. Park, E. I. Iwuoha and M. R. Smith, Electroanalysis, 1997, 9, 1120 CAS.
- P. Dominguez-Sanchez, C. K. O’Sullivan, A. J. Miranda-Ordieres, P. Tunon-Blanco and M. R. Smyth, Anal. Chim. Acta, 1994, 291, 344 CrossRef CAS.
- J. Wang and J. Liu, Anal. Chim. Acta, 1993, 284, 385 CrossRef CAS.
- N. Sulcova, I. Nemc, K. Waisser and H. L. Kies, Microchem. J., 1980, 25, 551 CrossRef CAS.
- R. Singh and R. A. Singh, Mol. Mater., 1998, 9, 343 Search PubMed.
- M. H. Bickel, in The Phenothiazines and Structurally Related Drugs, ed. I. S. Forrest, C. J. Carr and E. Usdin, Raven Press, New York, 1974, pp. 163–166. Search PubMed.
Footnote |
| † Presented at SAC 99, Dublin, Ireland, July 25–30, 1999. |
| This journal is © The Royal Society of Chemistry 2000 |